Что вы знаете о своей наследственности? [Николай Дмитриевич Тарасенко] (fb2) читать онлайн
- Что вы знаете о своей наследственности? (а.с. АН СССР. Научно-популярная литература. Человек и окружающая среда) 679 Кб, 101с. скачать: (fb2) - (исправленную) читать: (полностью) - (постранично) - Николай Дмитриевич Тарасенко - Галина Ивановна Лушанова
[Настройки текста] [Cбросить фильтры]
[Оглавление]
Н. Д. Тарасенко, Г. И. Лушанова Что вы знаете о своей наследственности?
АКАДЕМИЯ НАУК СССР СИБИРСКОЕ ОТДЕЛЕНИЕ 2-е издание, исправленное и дополненное Ответственный редактор доктор медицинских наук М. Н. Кириченко Рецензенты доктора биологических наук И. Ф. Жимулев, А. Н. Мосолов Утверждено к печати Центральным сибирским ботаническим садом СО АН СССР«Биография» книги Н.Д. Тарасенко, Г.И. Лушановой «Что вы знаете о своей наследственности?» Первое издание книги, вышедшее в Сибирском отделении издательства «Наука» в 1980 г. (тираж 50 тыс. экз., количество заявок 380 тыс), отмечено первой премией Всесоюзного общества генетиков и селекционеров им. Н.И. Вавилова и переведено на японский, китайский и узбекский языки. Второе, сокращенное, издание книги, вышедшее в издательстве «Медицина» в 1984 г., также имело большой спрос читателей (тираж 200 тыс, экз., количество заявок около 4 млн). Третье, настоящее, издание переработано и дополнено новыми данными, полученными за последние 10 лет.
 ТАРАСЕНКО Николай Дмитриевич -доктор биологических наук, специалист по генетике и цитологии, автор и соавтор более 200 научных работ, в том числе пяти монографий, и ряда научно-популярных книг. Более 30 его научных работ переведены на иностранные языки. Научная и общественная деятельность Н.Д. Тарасенко отмечена юбилейными медалями им. В.И. Ленина и им. Н.И. Вавилова.
ТАРАСЕНКО Николай Дмитриевич -доктор биологических наук, специалист по генетике и цитологии, автор и соавтор более 200 научных работ, в том числе пяти монографий, и ряда научно-популярных книг. Более 30 его научных работ переведены на иностранные языки. Научная и общественная деятельность Н.Д. Тарасенко отмечена юбилейными медалями им. В.И. Ленина и им. Н.И. Вавилова.
 ЛУШАНОВА Галина Ивановна — научный сотрудник Клинического центра Новосибирского института биоорганической химии СО АН СССР, специалист по генетике и цитологии, автор и соавтор более 30 научных публикаций. Ее главный научный интерес — изучение «поведения» лекарств в организме человека.
ЛУШАНОВА Галина Ивановна — научный сотрудник Клинического центра Новосибирского института биоорганической химии СО АН СССР, специалист по генетике и цитологии, автор и соавтор более 30 научных публикаций. Ее главный научный интерес — изучение «поведения» лекарств в организме человека.
Предисловие
Современная биология, особенно ее фундаментальные разделы, касающиеся основ организации и воспроизведения живых организмов, стремительно развивается. Столь же быстро прогрессируют генетика человека и медицинская генетика. На XIV Международном генетическом конгрессе в Москве (август 1978 года) упоминалось о более 2500 типах наследственных заболеваний, описанных главным образом за несколько предшествующих форуму десятилетий. Заболевания делятся на три большие группы — генные (ошибки в генах), хромосомные (нарушения числа и структуры хромосом) и тератогенные (повреждения эмбриона в период развития беременности). Установлено, что около 4,5—5% детей рождается наследственно неполноценными (отягощенными). Однако за последние 10—12 лет количество известных наследственных заболеваний увеличилось и составляет в настоящее время около 4000 наименований. Иными словами, каждый год выявляется примерно 100 новых заболеваний. Это происходит из-за того, что, с одной стороны, наука все более проникает в генетические, физиологические и биохимические механизмы человека, а с другой — экологическая среда обитания все в большей степени становится загрязненной и матрицы человека активнее подвергаются воздействию, нарушаются. Многие генетические заболевания (около 500) ученые научились «исправлять» или вести профилактику их посредством дието-, ферменто- и гормонотерапии с последующей генетической консультацией вступающих в брак. За последние годы в нашей стране издано много специальных книг по генетике человека. Это — «Основы генетики человека» К. Штерна, «Введение в медицинскую генетику» В. П. Эфроимсона, «Цитогенетика человека», «Лекции по медицинской генетике» и ряд брошюр — «О наследственности: хромосомы и хромосомные болезни» И. Исаевой, «Наследственность человека» и «Генетика человека» Б. В. Конюхова и Ю. В. Пашина и др. Однако для широкого круга читателей эта литература сложна, а научно-популярной, где бы просто и ясно излагались основные положения генетики человека, явно недостаточно. Настоящая книга частично восполняет этот пробел. Один из авторов (Н. Д. Тарасенко) уже более четверти века читает лекции на курсах усовершенствования учителей, слушателей Высшей партийной школы (Новосибирск), областных и межобластных курсах о проблемах и успехах генетики микроорганизмов, растений, животных и человека. Его опыт показывает, что большинство вопросов слушателей так или иначе связано с генетикой человека — наследованием групп крови системы АВ0, резус-фактором, генными и хромосомными болезнями и причинами их появления[1]. Авторы настоящей книги попытались ответить на наиболее часто встречающиеся вопросы. Прежде всего рассказывается о клетке как основе всего разнообразия жизни. Рассмотрены такие вопросы, как код наследственности, синтез (сборка) белков, деление клетки. Описаны довольно частые случаи отклонений в наборе хромосом, возникающих в период созревания половых гамет, в результате оплодотворения которых формируются дети с тяжелейшими заболеваниями. Это случаи рождения девочек с набором хромосом (кариотипом) мальчика, аномалиями половых хромосом, нарушениями неполовых хромосом (аутосом). Рассмотрены причины рождения детей с 47 хромосомами (на примере синдрома Дауна). Показана зависимость возраста матери и частоты аномалий нервной системы у детей. Частота хромосомных аномалий среди новорожденных высокая и составляет 1,5—2 %. Повествуется о наиболее распространенных генных «матричных» заболеваниях у детей, причинах их возникновения, о характере наследования (доминантные и рецессивные), сцепления с половыми хромосомами (локализованными в половых хромосомах) и др. Небольшой раздел посвящен принципу обнаружения генных заболеваний. Рассказывается о третьей группе врожденных аномалий, названных «тератогенными», то есть о заболеваниях, вызванных различными факторами (вирусами, лекарствами, курением, алкоголем и т. д.) в период формирования эмбриона. Эта группа составляет около 2,5—3,0 % новорожденных. Кроме того, значительное количество повреждений новорожденных происходит при рождении детей так называемыми старыми первородками — женщинами, рожающими первый раз в возрасте старше 25 лет. Книга содержит современные статистические данные о рождаемости во многих промышленно развитых странах, в том числе и в СССР. Показана четкая зависимость между количеством детей в семье и развитием коллективистских черт их характера. В семьях с одним или даже двумя детьми, родившимися с разрывом в 8—12 лет, часто формируются личности с эгоистическими наклонностями, что в социологической литературе получило название «эффект одиночки». В этой связи нельзя не вспомнить выдающегося советского педагога А. С. Макаренко, который считал оптимальной семью, имеющую трех детей, родившихся с небольшим временным интервалом, что позволяет им оптимально формироваться в микроколлективе. В книге рассматривается особая, невосполнимая значимость первых трех лет воспитания в развитии человека. Подчеркивается важность этого периода в становлении его характера, формировании основы интеллекта. Книга написана живым, доступным языком и читается с интересом. Авторам в основном удалось донести до читателей сложные и вместе с тем важные достижения и проблемы генетики человека. Представленная публикация, без сомнения, будет способствовать повышению знаний человека с самом себе. М. Н. КириченкоГлава 1. Клетка как основа жизни
Роль и синтез белков
Все многообразие живых организмов состоит из клеток. Клетка — это структурная и функциональная единица всего живого, производящая (строящая, синтезирующая) белки. Точное число белков, необходимое для нормальной жизнедеятельности клетки, неизвестно, но установлено, что оно достигает многих сотен тысяч. Нельзя найти двух людей, за исключением однояйцевых близнецов, имеющих полностью одинаковые белки. Белки состоят из 20 аминокислот: аланин, глицин, глютаминовая кислота, гистидин, глютамин, аспарагин, валин, лейцин, изолейцин, метионин, аргинин, лизин, треонин, тирозин, триптофан, фенилаланин, аспаргиновая кислота, серии, пролин и цистеин. Каждая молекула белка представляет собой цепь чередующихся в определенном порядке вышеперечисленных аминокислот, число которых может достигать шести — семи сотен. В организме человека белки выполняют разнообразные функции. Из белков и их составных частей формируются ферменты, которые способствуют осуществлению множества реакций, протекающих в каждой клетке. Белки могут действовать и как гормоны. При недостатке тех или иных аминокислот и белков отдельные жизненно важные химические реакции в организме могут быть приостановлены или даже выключены из обменных процессов, что приводит к нарушению обмена веществ и в итоге — к болезням. Полное лишение белковой пищи неизбежно приводит к смерти, даже при обильном питании жирами и углеводами. Белки различных пищевых продуктов неравноценны по содержанию в них аминокислот. Из 20 аминокислот для синтеза белков не все одинаково нужны. При недостаточном поступлении одних аминокислот белки могут быть частично синтезированы из других. Это так называемые заменимые аминокислоты. Однако девять аминокислот (из 20) должны поступать в организм обязательно, так как они не способны синтезироваться из других аминокислот. Это незаменимые аминокислоты. Среди последних наиболее часто используются лизин, метионин и триптофан. Они получили название критических. В опытах над животными установлено, что пища без незаменимых аминокислот очень скоро вызывает признаки белковой недостаточности — задержку роста, малокровие, выпадение волос и др. В каждой молекуле белка аминокислоты чередуются в определенной последовательности. Замена одной аминокислоты другой приводит к изменению структуры и функции молекулы белка, поэтому для организма такая замена не проходит бесследно. Например, молекула гемоглобина состоит из четырех белковых цепочек. Замена в каком-нибудь определенном месте в одной из цепочек, состоящей из 146 аминокислот, глютаминовой аминокислоты валиновой приводит к тяжелейшему заболеванию — серповидно-клеточной анемии (формируются эритроциты серповидной формы, не способные переносить кислород). Последовательность аминокислот при синтезе, сборке белков в клетках организма определяется генетической программой, заложенной в каждом организме. Эта программа хранится в ядре клетки в особых структурах, называемых хромосомами, в виде дезоксирибонуклеиновой кислоты (ДНК)[2]. Основной структурный материал хромосом — не чистая ДНК, а дезоксирибонуклеопротеид (ДНП), то есть комплекс, состоящий из белка и ДНК. Это химическое соединение было открыто еще в 1869 году, но только в 1948—1949 годах удалось доказать, что оно является молекулярным носителем наследственности. Рис. 7. Химическое строение азотистых оснований нуклеиновых кислот.
Рис. 7. Химическое строение азотистых оснований нуклеиновых кислот.
Молекула ДНК состоит из двух цепочек, подобных друг другу, закрученных в спираль. Каждая из цепочек представлена, в свою очередь, четырьмя чередующимися элементами — нуклеотидами. Нуклеотиды состоят из фосфатного остатка, соединенного с сахаром дезоксирибозой и одним из четырех азотистых оснований: аденином (А), гуанином (Г), тимином (Т) и цитозином (Ц), которые соединены с фосфорной кислотой через сахар — дезоксирибозу (рис. 1). Две цепочки в молекуле ДНК соединены водородными связями, которые образуются между парой оснований: аденин — тимин и гуанин — цитозин. Если в одной цепочке стоит основание А, то во второй, напротив, находится обязательно Т (рис. 2). Примерно 1000 чередующихся пар оснований соответствуют одному гену, а в каждой клетке содержится до одного миллиона генов. Совокупность всех генов представляет генотип организма, а реальное выражение последнего в человеке называется фенотипом. Фенотип в значительной мере зависит от реальных условий, в которых функционирует генотип. Если вытянуть в одну нить все ДНК из одной клетки человека, то ее длина составит 3 м 60 см. Во время митоза (процесса деления клетки) вся ДНК укладывается в хромосомы (хромо — цвет, сома — тело). Хромосом всего 23 пары, то есть 46 штук, и общая длина их не превышает 200 мкм.
 Рис. 2. Модель строения дезоксирибонуклеиновой кислоты (ДНК).
Рис. 2. Модель строения дезоксирибонуклеиновой кислоты (ДНК).
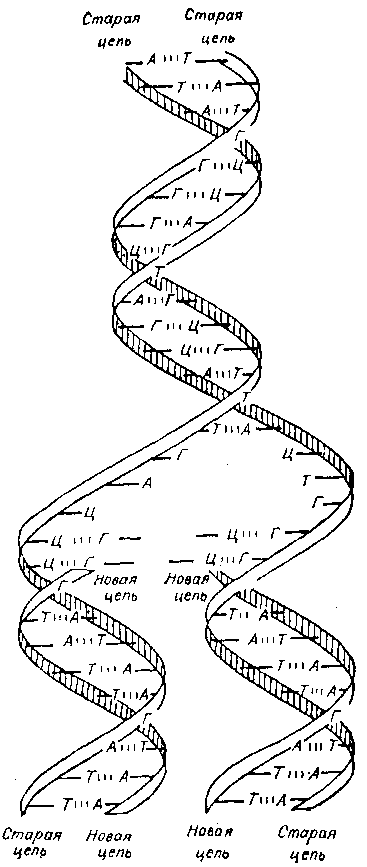 Рис. 3. Схема репликации ДНК.
Рис. 3. Схема репликации ДНК.
Одно из важнейших свойств ДНК — ее способность к самовоспроизводству, названная репликацией. При определенных условиях в клетке происходит репликация пар А — Т и Г — Ц. Две нити ДНК расходятся, причем на каждой из них, как на матрице, сразу же синтезируются аналогичные нити (рис. 3). Сама ДНК непосредственного участия в синтезе белка не принимает, однако она влияет на этот процесс опосредованно, через другую кислоту — рибонуклеиновую (РНК). В отличие от ДНК РНК одноцепочна, вместо сахара дезоксирибозы имеет рибозу и вместо основания тимина — урацил (см. рис. 1). В синтезе белка принимают участие три типа РНК: информационная (синтезируется на ДНК, как на матрице, в ядре клетки, затем покидает ядро и направляется к определенным структурам цитоплазмы — рибосомам, на которых осуществляется синтез белка); рибосомальная и, наконец, транспортная (доставляет определенные аминокислоты к рибосомам для сцепления их в молекулу белка). Каким же образом последовательность четырех оснований (А, Г, Т и Ц) в ДНК определяет (кодирует) последовательность 20 аминокислот в белке? Для этого природа в процессе эволюции создала так называемый генетический код. Как азбукой Морзе (тире с точкой) можно записать и передать любое сообщение, так и генетическим кодом, выраженным четырьмя основаниями в виде триплетов, можно записать последовательность каждой из 20 аминокислот и поставить их в определенное место в белковой цепочке — молекуле. Генетический код, открытый в 1961—1964 годах, оказался именно триплетным, то есть три нуклеотида в строго определенной последовательности кодируют свою аминокислоту в белке при его создании на специальной матрице — информационной РНК. Триплет — это не просто случайная группировка из трех нуклеотидов: каждый триплет определяет (кодирует) включение только своей аминокислоты. Установлено, что восемь аминокислот могут быть закодированы в среднем двумя разными триплетами, пять аминокислот — четырьмя, а три аминокислоты — аргинин, серин и лейцин — даже шестью триплетами, и только две аминокислоты имеют по одному кодирующему триплету (рис. 4).
 Рис. 4. Словарь генетического кода.
Рис. 4. Словарь генетического кода.
Способность кодировать (устанавливать на определенное место) одну и ту же аминокислоту разными триплетами получила название вырожденности генетического кода. Благодаря последней природе удается как бы снять шумы (возможные ошибки), возникающие при работе генетического материала, особенно при его удвоении. Из-за вырожденности (повторенности) генетического кода не каждое изменение оснований в триплетах может отражаться на последовательности и наборе аминокислот в белке, то есть изменять генетический смысл. Явление вырожденности генетического кода снижает частоту возможных спонтанных (естественных) и экспериментальных изменений (мутаций) на 24,5 %. Наличие вырожденности генетического кода является, можно сказать, непреодолимым барьером на пути получения наследственных изменений определенных признаков (локусов) по многим генам. Генетический код — удивительно универсальное явление, присущее всем известным организмам от бактериофагов до человека. Это подтверждает общность (из одного источника) происхождения всего живого, в том числе и человека.
Деление клетки
При рассмотрении клетки в обычный световой микроскоп видно, что границы ее, благодаря наличию оболочки, четко очерчены. Часто в клетках заметно ядро (рис. 5, 6), также имеющее оболочку. В некоторых клетках ядра не видно, но различаются структуры, названные хромосомами. Основной структурный материал хромосом (90 % массы) — дезоксирибонуклеопротеид (ДНП), то есть комплекс, состоящий из белка и ДНК. Отдельные участки хромосом, ответственные за проявление определенных признаков, называются генами. Рис. 5. Внешний вид клетки.
Рис. 5. Внешний вид клетки.
Хромосомы есть во всех клетках без исключения. Однако наблюдать их с помощью микроскопа можно только в том случае, если они сжаты, спирализованы, плотно упакованы. В период деления клетки ядерная оболочка растворяется, а хромосомы укладываются в спираль, хорошо окрашиваются специальными красителями и становятся видимыми в световой микроскоп. Далее хромосомы располагаются по экватору клетки, делятся и расходятся к разным полюсам. Делится и цитоплазма. Хромосомы вновь деспирализуются (раскручиваются) и образуют ядерную оболочку. Так из одной материнской клетки образуются две дочерние, совершенно одинаковые. Интересно, что количество ДНК в новых клетках не уменьшается, а остается прежним, так как до начала деления происходит его удвоение (репликация). Такое удвоение получило название митоза.
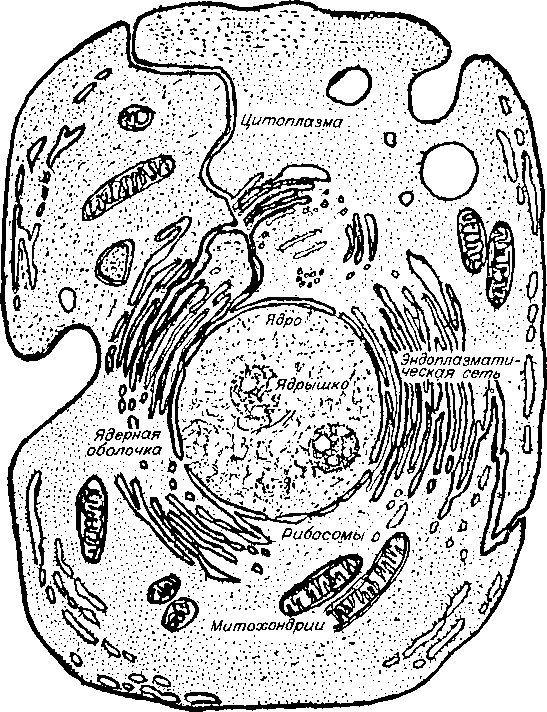 Рис. 6. Внутреннее строение клетки (схема).
Рис. 6. Внутреннее строение клетки (схема).
Хромосомы в клетках обнаружены учеными давно, однако только в 1902—1935 годах Томасом Г. Морганом и представителями его школы сформулирована хромосомная теория наследственности. Известно, что у одного и того же вида животных и растений количество хромосом одинаково во всех клетках (кроме половых), у разных же видов — различно. Так, у мыши их 40, крысы — 42, лисицы — 34, свиньи — 38, кролика — 44, человекообразной обезьяны — 48, осла — 62, лошади—64, дрозофилы — 8. Только в 1956 году было точно установлено, что у человека в клетках содержится 46 хромосом — 44 аутосомы и 2 половые хромосомы, а до этого времени считалось, что их 48, как и у обезьяны. Точный анализ хромосом удалось провести благодаря тому, что наука обогатилась новыми методами приготовления препаратов. Уже в 1959 году была выявлена хромосомная аномалия у человека — так называемый синдром Клайнфельтера. Эта болезнь была описана врачом еще в 1942 году. Ее характерные признаки: высокий рост, гинекомастия, атрофия яичек, мягкая форма дебильности и др. Причина появления этого синдрома — наличие лишней Х-хромосомы в генотипе больного (44 + + XXY). Интересно, что в 1949 году М. Барр обнаружил в ядре неделящейся клетки присутствие интенсивно красящегося объекта, который был назван именем ученого — тельцем Барра (половой хроматин). Последнее присутствует только в клетках женщин и отсутствует в клетках здоровых мужчин. Позднее было установлено, что при наличии двух Х-хромосом в клетке одна из них находится в плотно сжатом состоянии, образуя тельце Барра. У мужчин с синдромом Клайнфельтера в ядрах клеток также присутствует тельце Барра. В каждой клетке организма человека или животного имеются две хромосомы одного размера и одинаковой формы. Одна из них (гомологичная) получена or отца, другая — от матери. Чтобы число хромосом не возрастало от одного поколения к другому, в половых клетках (гаметах) их должно быть вдвое меньше, чем в зиготе (оплодотворенной яйцеклетке). Уменьшение же числа хромосом вдвое происходит в результате особого клеточного деления — мейоза, наблюдающегося при образовании гамет. При мейозе каждая из хромосом удваивается, гомологичные хромосомы сближаются, образуя пары. Этот процесс носит название конъюгации хромосом. Хромосомы вытягиваются (деспирализуются), что обеспечивает тесное сближение их отдельных участков. При этом в некоторых местах происходит перекручивание хромосом, составляющих пару. Затем, вследствие спирализации, конъюгирующие хромосомы укорачиваются, располагаются по экватору клетки и в анафазе (стадии деления ядра) сближенные ранее гомологичные удвоенные хромосомы расходятся к разным полюсам. Таким образом, к каждому полюсу отходит лишь одна из парных гомологичных хромосом. Обычно вслед за этим сразу начинается второе деление. Однако у человека в отличие от животных и растений эти два деления в значительной степени разделены во времени: первое редукционное деление хромосом (уменьшительное) плода происходит в период 3—6 месяцев внутриутробного развития, второе — спустя 10—12 лет (а последней половой клетки — примерно через 40 лет). Итак, в отличие от обычного деления (митоза) в мейозе ядро делится на два ядра, а хромосомы удваиваются один раз. В результате этих делений из одной клетки образуется четыре, число хромосом в которых уменьшается вдвое. Новые клетки содержат не двойной (диплоидный — 2п), а одинарный (гаплоидный — 1n) набор хромосом (рис. 7). При слиянии двух гаплоидных гамет в зиготе диплоидный набор хромосом восстанавливается. Сколько отцовских и сколько материнских хромосом получит каждая зигота? Это очень важно, так как оказывается, что хромосомы, полученные от отца и матери, рекомбинируются (обмениваются участками) в процессе мейоза совершенно свободно. При расхождении гомологичных хромосом к одному полюсу могут отойти две материнские, к другому — две отцовские. Однако с равной вероятностью могут состояться и другие комбинации — например, к каждому полюсу отойдут одна материнская и одна отцовская хромосомы. А если у человека 23 пары хромосом, то сколько же разнообразнейших комбинаций может возникнуть в гаметах? И каждый участок хромосомы (ген) оказывает специфическое влияние на развитие наследственных признаков организма. Таким образом, именно мейоз обеспечивает возникновение огромного разнообразия сочетания признаков родителей и потомков.
 Рис. 7. Мейоз и образование сперматозоидов (а) и яйцеклетки (б) у человека (схема).
Рис. 7. Мейоз и образование сперматозоидов (а) и яйцеклетки (б) у человека (схема).
Это разнообразие увеличивается еще и тем, что в процессе конъюгации гомологичные хромосомы обмениваются участками, наследственные особенности которых не всегда одинаковы. Первоначальное предположение о каком-то определенном расположении генов в хромосомах возникло тогда, когда на модельных объектах было установлено, что некоторые признаки, обусловленные генами, наследуются связанно друг с другом. Тенденцию признаков наследоваться совместно, а не порознь назвали сцеплением. Групп сцепления столько, сколько пар хромосом у конкретного вида. Ученые, тщательно изучив закономерности появления различных признаков при гибридизации у животных и растений, обнаружили, что сцепление признаков характерно как для животных (в том числе человека), так и для растений. В результате анализа сцепления и связанного с ним процесса обмена участками конъюгирующих хромосом в каждой паре (одна хромосома — от отца, другая — от матери) установлено, что гены в хромосомах расположены в линейном порядке. В настоящее время не только подтверждено линейное расположение генов в хромосомах, но и выяснена их сложная химическая структура в виде огромных молекул ДНК. Сейчас принято считать, что ген — это линейная последовательность пар нуклеотидов (от нескольких сотен до тысячи и даже более), кодирующая определенную функцию, а хромосома — это линейная последовательность генов. Основные закономерности наследования признаков описаны еще в 1865 году Грегором Менделем и основаны на расхождении хромосом в мейозе. Поскольку мейоз характерен для всех организмов, размножающихся половым путем, то закономерности наследования у них одни. Однако вернемся к митозу. Мы выяснили, что наследство распределяется наследницам поровну. В то же время при делении клетки и репликации (удвоении) генетического материала, хотя и редко, но происходят ошибки. Более того, именно путем проб и ошибок шла вся эволюция живых организмов. Именно ошибкам — отклонениям от генетической программы развития, которые происходят всегда и на всех уровнях жизни, обязан прогресс организмов, видов, родов, семейств. Некоторые отклонения от нормы как бы сообщают данному организму дополнительные возможности, а, значит, и некоторые преимущества перед другими организмами. Изменения, происходящие в наследственных структурах (ДНК), влияют на развитие и проявление новых признаков, морфологических, физиологических или биохимических особенностей.
Ошибки в наследственной программе — мутации
Наследственные изменения (мутации) организма обусловлены разными темпами изменений генетического материала независимо от рекомбинации генов в мейозе. Мутации бывают генными, хромосомными и геномными. Первые возникают в результате выпадения одного основания в молекуле ДНК, вставки дополнительного основания и замены одного другим. В настоящее время твердо установлено, что генные мутации происходят в периоды удвоения структуры ДНК (репликации) и работы генного материала, когда снимается копия с генов или целого блока генов (транскрипция). В этом случае двойная спираль ДНК также локально расплетается. Именно в процессе репликации и транскрипции основания (нуклеотиды) оказываются наиболее доступными для химических соединений, которые модифицируют их, что и приводит к ошибкам репликации, или, как их еще называют, ошибкам спаривания. В результате меняется очередность нуклеотидов в ДНК дочерних клеток по сравнению с материнской. Хромосомные мутации также могут быть нескольких видов: дупликации (добавление родственного участка хромосомы); делеции (выпадение участка); инверсия (поворот какого-либо участка внутри хромосомы на 180 °) и транслокации (обмен негомологичных хромосом отдельными участками). Существуют хромосомные мутации и другого типа — это тот случай, когда организм отличается от нормы одной или несколькими хромосомами. Такое явление получило название гетероплоидии. Геномные мутации возникают вследствие кратного изменения наборов или геномов хромосом. Таким образом, одни мутации приводят к изменению последовательности расположения нуклеотидов, другие обусловливают наличие неодинакового количества хромосом во вновь образованных клетках, третьи нарушают нормальную работу митотического, или мейотического, деления, что блокирует расхождение отдельных пар хромосом по дочерним клеткам. Частота появления перечисленных типов мутаций разная и очень низкая. Она составляет в среднем одну мутацию на миллион клеток. Однако если учесть огромнейшее количество клеток, составляющих организм человека, то в целом частота мутирующихся генов значительна. Разумеется, для того чтобы быть переданными следующему поколению, мутации должны произойти не в обычных клетках (соматических), а в половых. Теперь, когда мы заглянули во внутрь клетки, интересно проследить за изменениями в генетической программе, в очередности триплетов в ДНК под действием химических и физических факторов (разные виды ионизирующей радиации). Подсчитано, что для разрыва хромосомы толщиной 0,2 мк необходимо, чтобы на пути пробега через нее альфа-частицы образовалось около 15—20 пар ионов. Следует отметить, что степень изменения хромосомы зависит от места, через которое проходит ионизирующая частица, то есть от того, какой ген претерпевает изменение. Возникает вопрос: будет ли изменение в ДНК конкретного гена гарантией мутации. Оказывается, что нет. Исследования показали, что на пути репликации имеется целый ряд барьеров, только с преодолением которых мутация от первичного изменения в ДНК может выйти на фенотип. В последние годы учеными обнаружен сложный барьер в виде системы восстановления, стоящий на пути возможного нарушения очередности оснований в генах. Установлено, что если облучить бактериальные клетки (например, кишечную палочку) или обработать их специальными химическими веществами, вызывающими изменение очередности оснований в молекулах ДНК, то через некоторое время примерно 40—60 % участков ДНК, несущих повреждения, может бить «вырезано» специальными ферментами и заново «застроено» (как бы отремонтировано) комплементарными основаниями. Это происходит потому, что по неизмененной цепочке ДНК клетка способна полностью восстанавливать исходную двойную структуру своего генного материала. В результате сложных процессов, происходящих в клетке после облучения ионизирующей радиацией или воздействия химическими веществами, возникает новая последовательность расположения пар оснований. Однако эта мутация пока не связана с генетическими последствиями. Клетке необходимо преодолеть еще ряд преград, чтобы изменение на молекулярном уровне могло бы реализоваться на уровне организма. Предположим, что один барьер преодолен, и поврежденные участки восстановлены лишь частично. В результате появился новый порядок (очередность) оснований в ДНК. Приведет ли это к изменению признаков организма? Нет, не приведет. Следующей преградой является вырожденность генетического кода, или, как ее еще называют, повторяемость, которая также значительно снижает частоту вызванных мутаций. Далее. Известно, что из четырех оснований при составлении всех возможных триплетов получается 64 комбинации, а наиболее часто встречающихся аминокислот — только 20. Ученые воссоздали все 64 типа триплетов экспериментально, а генетический код при этом оказался триплетным. Последнее означает, что три нуклеотида кодируют (устанавливают) конкретную аминокислоту в определенное место в полипептидной цепи при создании молекулы белка на специальной матрице — информационной РНК. Возникает вопрос. Почему в процессе эволюции природа так жестко унифицировала генетический код, а не сохранила для каждой аминокислоты свой уникальный, единственный, триплет (см. рис. 4)? Оказывается, эволюция произвела такую унификацию для сохранения стабильности в работе генетического материала и устранения возникающих помех (ошибок), как бы своего рода шумов. Природа поставила препятствия на пути изменения очередности оснований в ДНК, иными словами, создала запас прочности работы молекулярных основ жизни. Представим себе такую ситуацию. При облучении клеток или обработке их химическими мутагенами удалось изменить очередность нуклеотидов в гене таким образом, что вместо триплета, например, АГУ образовался один из триплетов УЦЦ, УЦА, УЦГ, УЦУ или АГЦ. В этом случае при сборке (синтезе) белка в нить информационной РНК встраивается та же аминокислота, какова была и у неизмененного, исходного, триплета — а именно, серия. Возникшая новая очередность нуклеотидов в гене не сможет реализоваться, то есть дойти до генетических последствий, из-за вырожденности генетического кода. Следовательно, не каждое изменение в очередности оснований в гене приводит к мутации. Таким образом, вырожденность генетического кода оказалась одной из гарантий стабильности работы генетического материала. Предположим теперь, что вызванное изменение в очередности оснований ДНК не было нейтрализовано вырожденностью генетического кода. Необходимо далее, чтобы это изменение не было задержано, снято соматическим (клеточным) отбором. Иными словами, необходимо, чтобы приобретенное клеткой изменение не сказалось на темпе деления, в противном случае такая клетка не сможет конкурировать с нормальными, неизмененными, клетками. Только в том случае, если клетка, в которой произошли изменения, будет нормально функционировать и размножаться, эти изменения будут переданы дочерним клеткам и смогут дойти до морфологического или физиобиохимического проявления. Однако часто такая клетка размножается слабее нормальной и не может успешно конкурировать с последней. В силу этого происходит клеточный (соматический) отбор, что приводит к снижению частоты наследственных изменений — клетка с изменениями будет просто вытеснена нормальными клетками. Однако допустим, что и этот барьер преодолен. Дойдет ли теперь полученное изменение до фенотипа? Чтобы это произошло, необходимо еще одно условие. Еще один своего рода барьер существует на уровне протекания мейоза в половых клетках, когда многие изменения, прошедшие первые три преграды, могут быть задержаны, как бы нейтрализованы. Итак, барьеры для мутации природа создала, однако наследственные болезни существуют и передаются из поколения в поколение. В чем же причина? В 1908 году математик из Англии Харди и врач из Германии Вайнберг независимо друг от друга, проведя математический анализ распределения генов в популяции, обнаружили закономерность, хорошо описывающуюся формулой, отражающей количественную сторону генотипов и фенотипов в популяции. По закону Харди — Вайнберга, свободно существующие популяции при любом соотношении аллелей из поколения в поколение сохраняют их концентрации постоянными. Но это справедливо для идеальных условий, а в реальной жизни количество аллелей постоянно изменяется. Последнее происходит при родственных браках, мутировании генов, отборе, а также при таких социальных явлениях, как изоляция, миграция и др. Более подробно изменение частоты генов в популяциях описано Н. П. Дубининым (1966, 1970, 1986 гг.) и В. П. Эфроимсоном (1968 г.). Процесс отбора по устранению нежелательных генов из популяции наиболее эффективен в том случае, когда он направлен против доминантных мутаций при условии их полного выражения и проявления (экспрессивность и пенетрантность). В качестве примера можно привести болезнь ахондроплазию (доминантная карликовость). Когда эта болезнь проявляется в более позднем возрасте и носитель летального гена уже имеет потомство, то, естественно, такой доминантный ген уже передан потомкам (например, хорея Гантингтона). Бороться с переходом этой и аналогичных болезней от данного поколения к следующим можно лишь избегая деторождении в этих семьях.Глава 2. Хромосомные аномалии
Хромосомные аномалии представляют собой изменение наследственного материала, включающего сотни генов, и связаны с изменением числа или строения хромосом. Наличие лишней хромосомы (трисомия) или отсутствие одной из них (моносомия) ведет к избытку или недостатку многих генов, что связано с изменением многих фенотипических признаков организма. Такие хромосомные аномалии (синдромы), как правило, называются по имени ученых, которые их открыли и описали. В 1960 году английский ученый Ноуэл создал новый метод получения препаратов хромосом из периферической крови. До этого времени все работы по изучению хромосом человека проводились на материале, получаемом в результате операций. В современных экспериментах для кариологических исследований используются кровь, костный мозг, кожа и другие ткани, полученные разными методами. При этом кровь чаще всего берется из вены, пальца, пуповины (при ее обрезе у только что родившихся), а у детей до одного года — в основном из пятки.Мужские и женские хромосомы
Как уже упоминалось, в клетках человека содержится 46 хромосом, собранных в 23 пары (в каждой паре по одной хромосоме от каждого родителя), которые, в свою очередь, условно делятся на блоки А — G. Двадцать две пары называются аутосомными. Они одинаковы по размеру и обязательно присутствуют во всех клетках как мужчины, так и женщины. В двадцать третьей паре хромосомы не одинаковы по размеру. Эти хромосомы определяют пол в поэтому называются половыми. Генотип женщины содержит две хромосомы XX, а мужчины — XY (рис. 8). Одну из них, крупную, называют Х-, а другую, меньшую,— Y-хромосомой. Формы и размеры хромосом человека различны. Каждая из них имеет перетяжку (центромеру), которая делит хромосому на две части (плеча). В зависимости от расположения центромеры различают три типа хромосом: метацентрические (центромера расположена в центре хромосомы и оба плеча хромосомы одинаковы по длине); субметацентрические (центромера расположена не в центре хромосомы, одно плечо несколько больше другого); акроцентрические (центромера расположена на конце хромосомы, одно плечо длинное, а другое очень короткое). Рис. 8. Хромосомы женщины (а) и мужчины (б). Справа внизу — общий вид под микроскопом.
Рис. 8. Хромосомы женщины (а) и мужчины (б). Справа внизу — общий вид под микроскопом.
Поскольку размеры хромосом человека различны — есть очень большие и очень маленькие, их условно объединяют в шесть групп, в каждую из которых входят хромосомы, подобные по форме и размеру. До последнего времени было довольно трудно определять (идентифицировать) хромосомы внутри каждой группы. Об этом может свидетельствовать следующий пример. Опытным цитогенетикам предложили для идентификации препараты хромосом от одних и тех же лиц. В препаратах метафазных хромосом от нормальных мужчин в 22 % случаев определение оказалось неверным, а в препаратах от мужчин с различными аномалиями хромосом число правильных определений колебалось от 31 до 70 % Позднее был предложен флуоресцентный метод окраски хромосом с использованием красителей — атебрина, акрихин-иприта и др. Этот метод особенно успешно применяется для идентификации Y-хромосомы, которая интенсивно светится в метафазных и интерфазных пластинках (неделящихся клетках) и в сперматозоидах. В настоящее время при аномалиях половых хромосом наряду с другими методами идентификации хромосом считается необходимым применение метода флуоресцентной окраски Y-хромосомы.
Женщины с хромосомами мужчин
Флуоресцентный метод окраски Y-хромосомы подтвердил наличие мужских 44 + XY-хромосом у женщин с синдромом тестикулярной феминизации. Необходимо отметить, что этот синдром описан в клинической медицине задолго до XX века и диагностировался чаще всего при тщательном обследования или при оперативном хирургическом вмешательстве. В настоящее время, когда стало известно, что внешне нормальные женщины — носительницы синдрома тестикулярной феминизации — имеют XY-, а не ХХ-хромосомы, возможен массовый скрининг (обследование) этого заболевания. Чаще всего носителей хромосомных аномалий обнаруживают врачи-педиатры среди детей, так как их внешний вид и развитие резко отличаются от нормального уровня. Однако этого нельзя сказать об указанном выше синдроме тестикулярной феминизации. Видимо, причина здесь в том, что в данном случае имеется сбалансированный и нормальный хромосомный набор мужчины. Объясняя причину возникновения этого синдрома, некоторые ученые считают, что в результате мутации одного из генов организм оказался нечувствительным к мужским половым гормонам и на каком-то этапе развития эмбриона началось развитие по женскому тину. Для женщин с тестикулярной феминизацией характерно отсутствие менструаций, поэтому их иногда диагностируют врачи-гинекологи. Для безошибочного диагноза, помимо клинической картины, необходим анализ, устанавливающий кариотип хромосом. Цитолог, обнаружив у женщины набор хромосом 44 + XY, может предположить синдром тестикулярной феминизации, но ставить диагноз после соответствующего обследования может только врач. Тем не менее установление кариотипа 44 + XY, например, у женщин-спортсменок — достаточный фактор для вывода их из состава соревнований, так как считается, что такая женщина в спорте обладает возможностями мужчины. Таким образом, женщины иногда могут иметь хромосомы мужчины. Эта аномалия хотя и редко, но встречается. Логично было бы предположить, что должна встречаться и обратная аномалия — мужчины с женским набором хромосом (44 + XX). Такие мужчины действительно были обнаружены и описаны. Встречаются и мозаики по половым хромосомам. У них в одних клетках одного и того же организма встречаются клетки с кариотипом 44 + XX, а в других - 44 + XY. В научной литературе высказаны две гипотезы, объясняющие развитие мужского фенотипа при кариотипе 46 XX (44 + XX). Согласно первой, происходит транслокация (перемещение) части Y-хромосомы на Х-хромосому; согласно второй, имеет место мозаицизм, то есть в одних клетках данного организма встречается кариотип 44 + XX, а в других — 44 + XY. По многочисленным данным, частота хромосомных аномалий и, как следствие этого, тяжелых заболеваний среди новорожденных составляет 1,5—2,0 %. Что касается более грубых нарушений хромосом, то они несовместимы о жизнью и являются причиной частых спонтанных абортов, выкидышей и мертворождений. Это имеет место в 25 % случаев. Как видим, частота хромосомных аномалий весьма значительна.Аномалии половых хромосом
К настоящему времени описаны различные аномалии половых хромосом. Это синдром Шерешевского — Тернера (кариотип состоит из 45 хромосом Х0), Клайнфельтера (47 хромосом XXY), трисомии X (47 хромосом XXX), тетрасомии X (48 хромосом ХХХХ) и другие, которые встречаются гораздо реже (табл. 1). Больные с синдромом Шерешевского — Тернера имеют характерный фенотип: наличие крыловидной шейной складки, низкий рост, задержка полового развития, аменорея, бесплодие. Сейчас известно, что ряд больных с этим синдромом представлен мозаиками. Так, например, описаны больные с набором хромосом 45Х/46Х (изохромосома) или 45Х/46ХХ. Частота синдрома Шерешевского — Тернера среди новорожденных небольшая (1 из 2500), однако среди спонтанно абортированных эмбрионов она значительна, так как при хромосомном наборе 44 + ХО эмбрионы имеют пониженную жизнеспособность.Таблица 1. Наиболее частые отклонения по набору половых хромосом
| Норма и аномалия | Фенотип по полу | Комплекс половых хромосом |
|---|---|---|
| Нормальный мужчина | Мужской | XY |
| Нормальная женщина | Женский | XX |
| Синдром Шерешевского — Тернера | // | Х0 |
| Синдром Клайнфельтера | Мужской | XXY |
| Трисомия X | Женский | XXX |
| Трисомия X + Y | Мужской | XXXY |
| Тетрасомия X | Женский | ХХХХ |
| Тетрасомия X + Y | Мужской | XXXXY |
Аномалии аутосом
В качестве примера аномалий аутосом чаще всего приводится синдром Дауна. Эта тяжелейшая болезнь встречается часто и в относительных цифрах составляет 1 из 500—600. Больной с названным синдромом имеет непропорционально маленькую голову, узкие глазные щели, плоское лицо и резко выраженные признаки умственной отсталости. Кроме того, у 40 % больных синдромом Дауна наблюдаются различные пороки сердца. Генетически все эти проявления связаны с наличием у больного одной лишней хромосомы — 21-й. Иными словами, пациент имеет во всех клетках, включая половые, не 46 хромосом, как в норме, а 47 (рис. 9).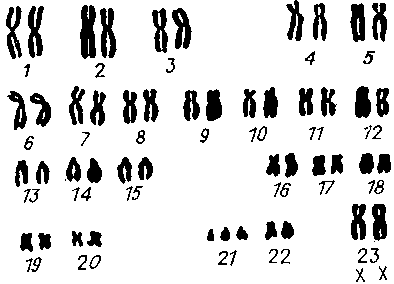 Рис. 9. Кариотип больного с синдромом Дауна с 47 хромосомами.
Рис. 9. Кариотип больного с синдромом Дауна с 47 хромосомами.
Если принять число всех больных с синдромом Дауна за 100%, то трисомия по 21-й хромосоме наблюдается у 94,5% пациентов, мозаицизм — у 2,27%, а синдром Дауна транслокационной природы — у 2,88 % пациентов. В последнем случае у больного имеются 46 хромосом, одна из них представлена объединением двух хромосом — чаще всего 15-й и 21-й (рис. 10). Обычно у одного из родителей, имеющих больного ребенка с синдромом Дауна, наблюдают 45 хромосом, при этом 15-я хромосома также объединена с 21-й. В такой семейной паре независимо от возраста матери дети с синдромом Дауна будут рождаться довольно часто. В случае синдрома Дауна, обусловленного мозаицизмом хромосом, при изучении кариотипа в культуре лимфоцитов (эта методика применяется в практике медико-генетической консультации) установлен очень низкий процент клеток с 47 хромосомами. В. Хаберландтом в 1973 году при исследовании кариотипа в культуре клеток с 47 хромосомами установлена трисомия по 21-й хромосоме в 100 % изученных клеток. В культуре лимфоцитов крови только две клетки из 103 имели кариотип с 47 хромосомами, а в культуре фибробластов кожи во всех клетках установлено 47 хромосом.
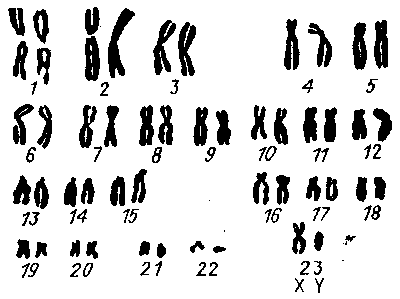 Рис. 10. Кариотип больного с синдромом Дауна с 46 хромосомами.
Рис. 10. Кариотип больного с синдромом Дауна с 46 хромосомами.
В литературе имеются сообщения о том, что причиной рождения детей с синдромом Дауна может быть мозаицизм одного или обоих родителей. В одном из таких описанных случаев в культуре лимфоцитов крови отца из 100 изученных клеток шесть имели 47 хромосом. Частота мозаиков с низким процентом 47-хромосомных клеток пока не известна. Почему же появляются дети с 47 хромосомами? И что происходит при образовании и формировании нового организма? Родители имеют по 46 хромосом и у ребенка (в норме) должно быть столько же. В половых клетках человека, образующихся в результате мейоза, содержится 23 хромосомы. У женщин при формировании половой клетки в последнюю попадает 23 хромосомы (22+ Х). Во всех яйцеклетках содержится только одна Х-хромосома. При формировании мужских половых клеток — сперматозоидов — в одну попадает Х-хромосома (22+ Х), а в другую — Y-хромосома (22 +У). Оплодотворение любой яйцеклетки, всегда имеющей хромосомы 22 + X, сперматозоидом с хромосомами 22 + X обусловливает развитие женского организма, а в случае оплодотворения сперматозоидом с хромосомами 22 + Y обеспечивается развитие мужского организма (рис. 11).
 Рис. 11. Передача половых хромосом от родителей к детям. Пояснения см. в тексте.
Рис. 11. Передача половых хромосом от родителей к детям. Пояснения см. в тексте.
Формирование половых клеток — очень сложный и ответственный процесс, протекающий в организме. Казалось бы, этот процесс хорошо защищен от воздействия различных факторов, однако, как показывает реальная жизнь, материнский возраст является важнейшим фактором, влияющим на возможность появления синдрома Дауна у ребенка. Установлено, что у 1000 женщин-рожениц в возрасте 15—19 лет частота появления новорожденных с синдромом Дауна крайне мала, и мы ее примем за единицу. У того же количества женщин в возрасте 35—39 лет частота рождения детей с синдромом Дауна будет гораздо большей и составит 11. После 45 лет эта цифра достигает уже 104 (табл. 2). Женщины старше 40 лет рожают примерно 2 % детей, однако среди новорожденных 24,5 % — то есть почти каждый четвертый — является обладателем синдрома Дауна. При этом вероятность появления на свет ребенка с синдромом Дауна не зависит от того, будет ли это первый ребенок в семье или последующий. Возраст матери не безразличен и для состояния нервной системы новорожденных, а именно — его увеличение повышает частоту врожденных аномалий. Если частоту нервных аномалий у детей, рожденных самыми молодыми (15— 19 лет) матерями, принять за единицу, то при возрасте рожениц 31—35 лет частота этих аномалий увеличивается в 4,3 раза, а при возрасте 41—45 лет —уже более чем в 100 раз! Таблица 2. Зависимость частоты рождения детей с синдромом Дауна от возраста матери *
| Возраст матери, лет | Сравнительная частота болезни Дауна |
|---|---|
| 15-19 | 1,0 |
| 20-24 | 1,3 |
| 25-29 | 1,3 |
| 30-34 | 3,6 |
| 35-39 | 11,0 |
| 40-44 | 41,3 |
| 45 и старше | 104,0 |
В настоящее время установлено, что первое редукционное деление (мейоз) у девочки происходит в эмбриональный период (4—6 месяцев беременности матери), а второе редукционное деление и созревание яйцеклетки наступают в период полового созревания, что соответствует примерно 11—12 годам. Далее попеременно, то в правом яичнике, то в левом, созревает одна яйцеклетка в каждые 28—30 дней. Таким образом, одни клетки будут «ждать» второго редукционного деления 11—12 лет, а другие — 25 или даже 40 лет. Однако на 100 женщин встречается одна, у которой созревают одновременно две и более яйцеклетки. Случаи рождения трех—пяти близнецов не единичны; имеются факты рождения шести близнецов. Во всех этих случаях близнецы разнояйцевые. В некоторых случаях созревающая и оплодотворенная яйцеклетка может поделиться и так, что даст возможность образоваться двум и более совершенно одинаковым (идентичным, однояйцевым) организмам-близнецам (рис. 12). Если разнояйцевые близнецы могут быть одно- или разнополыми, то однояйцевые бывают только однополыми. Частота рождения близнецов зависит от разных причин, в том числе и от возраста матери и числа ее предшествующих родов. Доказано, что с возрастом матери (табл. 3) и порядковым номером родов частота рождения близнецов увеличивается. Описаны такие семьи, в которых женщины имели близнецов в ряде поколений, то есть обладали наследственным предрасположением к многоплодию.
 Рис. 12. Однояйцевые сестры-близнецы.
Рис. 12. Однояйцевые сестры-близнецы.
Рождение близнецов одного пола еще не является доказательством их идентичности (однояйцевости). Однояйцевые близнецы, как правило, очень похожи друг на друга, однако случаются и исключения из этого правила. Последнее бывает в том случае, когда условия внутриутробного развития близнецов несколько отличались. А так ли важно знать, родились разно- или однояйцевые близнецы? Да, очень важно. Ведь если заболеет один из пары однояйцевых близнецов, врач должен пристально наблюдать и за другим, а, возможно, даже начать его профилактическое лечение. Изучение развития и заболеваемости близнецов оказало большое влияние на понимание роли наследственности и среды формирования человека в возникновении многих его болезней. Если какой-то признак имеет сходство у однояйцевых близнецов, то это является свидетельством его зависимости от наследственности (генотипа). Изучение заболеваемости одно- и разнояйцевых близнецов показало, что такие заболевания, как корь, коклюш, ветряная оспа и др., возникают только при наличии инфекции, а предрасположение организма к ним имеет малое значение. А вот для появления дифтерии, свинки, воспаления легких, полимиелита, туберкулеза и ряда других болезней, кроме первого фактора, играют роль и наследственные свойства организма.
Таблица 3. Возраст матери и частота рождения близнецов
| Возраст матери, лет | Частота рождения близнецов* |
|---|---|
| 18 | 27 |
| 20 | 36 |
| 25 | 62 |
| 30 | 90 |
| 35 | 48 |
| 40 | 25 |
| 45 | 73 |
| 48 | 30 |
Количество ежемесячно созревающих яйцеклеток находится под генетическим контролем и также сильно зависит от возраста матери. Совершенно естественно, что клетки, из которых формируются яйцеклетки, со временем стареют, то есть «портятся», и как бы природа их не защищала, они все равно с определенной вероятностью повреждаются. В результате этого второе редукционное деление может произойти с ошибками, например с нерасхождением хромосом. В таком случае будут формироваться яйцеклетки с 24 хромосомами, или вообще хромосомы распределятся по двум дочерним клеткам не, поровну, вследствие чего в яйцеклетку попадет больше хромосом, чем это необходимо. Оплодотворение такой клетки приведет к появлению ребенка с хромосомными аномалиями. Для объяснения причин, вызывающих появление хромосомных аномалий, ученые выдвинули ряд предположений. 1. Изменение pH (кислотности среды), наступающее в организме в результате старения, болезней и (особенно) воспалительных процессов половых органов женщин, что приводит к нерасхождению хромосом. 2. Изменение гормонального статуса (эндокринопатия). Последнее может привести к появлению детей с мозаицизмом хромосом и с синдромом Дауна. Напомним, что эндокринные отклонения с возрастом женщины увеличиваются. 3. Применение различных лекарств или использование воды и продуктов питания, содержащих химические вещества. В литературе имеются данные о связи между концентрациями фтора в воде и повышенной частотой рождения детей с синдромом Дауна (центральные штаты США). Так, при содержании 0,1 — 0,2 % фтора частота больных детей составляет 0,39 на 1000 новорожденных, а при 1,0—2,6 % — она возрастает до 0,58. В этом случае другой важный фактор, влияющий на частоту появления синдрома Дауна,— возраст матери, отходит на второй план. А ведь во многих странах вода фторируется для снижения заболеваемости кариесом зубов! И тяжесть последствий для потомства при этом, к сожалению, не учитывается! Как указывалось выше, у женщин формируется в среднем одна половая клетка в месяц. А это значит, что при оплодотворении совершенно отсутствует возможность выбора. У мужчин с возрастом количество половых клеток с неправильным набором хромосом каждые 15—18 лет увеличивается в 100 раз. Это, казалось бы, должно приводить к еще большей частоте новорожденных с синдромом Дауна. Однако природа распорядилась по-своему. Она «позволила» мужчине образовывать не одну половую клетку в месяц, а много миллионов, что создает возможность выбора, как бы конкуренции. Поэтому «вина» мужчин в появлении детей с синдромом Дауна (не транслокационной природы) гораздо меньше. В целом рождение детей с болезнью Дауна примерно в 80 % случаев обусловлено нерасхождением хромосом при формировании женских половых клеток и в 20 % — при формировании мужских половых клеток — сперматозоидов[3]. При этом нерасхождение хромосом может происходить как в первом делении в мейозе (когда будущая мать сама является эмбрионом), так и во втором (задолго до оплодотворения). Данные исследования материнского (отцовского) происхождения добавочных хромосом у больных с синдромом Дауна приведены в табл. 4. Интересен опыт анализа частоты рождения Даунов в Дании, где генетика человека хорошо поставлена на службу обществу. В 1950 и 1970 годах в этой стране был организован учет данных о рождении Даунов по возрастным группам рожениц (табл. 5). Оказалось, что в 1950 году из 79558 новорожденных 116 детей были с синдромом Дауна (в среднем 1 на 685 новорожденных). Спустя 20 лет, в 1970 году, из 70802 новорожденных обнаружен только 71 случай синдрома Дауна (в среднем 1 на 994 ребенка), то есть почти на 30 % меньше. Если проанализировать частоту рождения Даунов по возрастным группам рожениц, то видно, что за 20 лет возраст их уменьшился почти на 5 лет.
Таблица 4. Влияние пола родителей на частоту появления добавочной хромосомы у больных с синдромом Дауна
| Общее число обследованных больных | Число больных с добавочной хромосомой при первом и втором (в скобках) делении в мейозе | |
|---|---|---|
| Материнская хромосома | Отцовская хромосома | |
| 67 | 37 (7) | 14 (9) |
| 145 | 96 (16) | 20 (13) |
| Возраст матери, лет | Число детей с синдромом Дауна к общему числу новорожденных | 1950 г. | 1970 год | ||
|---|---|---|---|---|---|
| Число живорожденных | Ожидаемое число детей с синдромом Дауна | Число живорожденных | Ожидаемое число детей с синдромом Дауна | ||
| 15-19 | 1 : 2300 | 5 769 | 2,5 | 5 884 | 2,6 |
| 20-24 | 1 : 1600 | 22 310 | 13,9 | 26 588 | 16,6 |
| 25-29 | 1 : 1200 | 24 218 | 20,2 | 24 193 | 20,2 |
| 30-34 | 1 : 880 | 15 438 | 17,5 | 9 864 | 11,2 |
| 35-39 | 1 : 290 | 8 874 | 30,6 | 3 453 | 11,9 |
| 40-41 | 1 : 100 | 2 272 | 27,7 | 774 | 7,7 |
| 45-49 | 1 : 46 | 177 | 3,8 | 46 | 1,0 |
| Всего... | 79 558 | 116,2 | 70 802 | 71,2 | |
| Частота появления синдрома Дауна | 1 : 685 | 1 : 994 | |||
Глава 3. Генные заболевания у человека
Генная программа человека
Сколько генов у человека? Теоретические расчеты показывают, что у человека вся генетическая программа состоит примерно из 3,5 миллионов пар генов. К 1978 году описано около 3 тыс. генов и изучен характер их наследования. Оказалось, что 1489 генов — аутосомно-доминантные, 1117 — аутосомно-рецессивные и более 200 генов локализовано в Х-хромосоме (табл. 6). Конечно, изучено еще мало. Но в настоящее время ежегодно описывается до десяти новых генов. Для реализации свойств и функций организма «работают» все гены, но не одновременно. Здесь существует определенный жесткий порядок, последовательность, которая также находится под генетическим контролем. О том, что не все гены начинают «работать» сразу после рождения, свидетельствует следующий факт: примерно 1,5% новорожденных имеют генную отягощенность (неполноценность). В процессе жизни (онтогенеза) обнаружится еще довольно много заболеваний, связанных с неполноценностью генов, которые начинают «работать», «выйдя на фенотип» через генные продукты — информационные РНК, неполноценные ферменты и т. д. Откуда у человека появляются неполноценные, или так называемые летальные и полулетальные, гены? Человек прошел длительную эволюцию, прежде чем из одной клетки появился разумный, самый совершенный вид живой материи, могущий думать и изучать окружающий мир и себя. Естественно, что раньше появления биологической материи как новой формы в природе должны были возникнуть основания ДНК, из которых затем образовались матрицы, способные записать и передать следующему поколению признаки и свойства. Для всего этого потребовалось очень и очень много времени.Таблица 6. Динамика обнаружения наследственных (менделирующих) признаков у человека
| Тип наследования генов | 1958 г. | 1966 г. | 1968 г. | 1971 г. | 1975 г. | 1978 г. |
|---|---|---|---|---|---|---|
| Аутосомно-доминантный | 285 | 837 | 793 | 943 | 1218 | 1489 |
| Аутосомно-рецессивный | 89 | 531 | 629 | 783 | 947 | 1117 |
| Сцепленный с Х-хромосомой | 38 | 119 | 123 | 150 | 171 | 205 |
| Всего... | 412 | 1487 | 1545 | 1876 | 2336 | 2811 |
Таблица 7. Распространенность доминантных и аутосомно-рецессивных заболеваний в Европе (на 1000 новорожденных)
| Доминантные заболевания | |
|---|---|
| Наименование болезни | Частота, % |
| Хорея Гантингтона | 0,5 |
| Нейрофиброматоз | 0,4 |
| Миотоническая дистрофия | 0,2 |
| Множественный полипоз кишечника | 0,8 |
| Аплазия диафизов | 0,5 |
| Доминантная форма слепоты | 0,1 |
| Доминантный отосклероз (тип взрослых) | 1,0 |
| Гиперхолестеринемия моногенная | 2,0 |
| Несовершенный дентиногенез | 0,2 |
| Поликистоз почек (тип взрослых) | 1,0 |
| Туберкулезный склероз | 0,01 |
| Базилярные вдавления | 0,03 |
| Тинатоформная карликовость | 0,08 |
| Синдром Марфана | 0,04 |
| Ахондроплазия | 0,02 |
| Синдром Элерса — Даплоса | 0,01 |
| Остеопетроз | 0,01 |
| Ретинобластома | 0,03 |
| Расщепление губы и (или) неба со слизистыми ямками у губ | 0,01 |
| Несовершенный остеогенез | 0,02 |
| Аутосомно-рецессивные заболевания | |
| Наименование болезни | Частота, % |
| Муковисцидоз | 0,5 |
| Фенилкетонурия классическая | 0,1 |
| Нейрогенные мышечные атрофии | 0,1 |
| Серповидно-клеточная анемия | 0,1 |
| Гиперплазия надпочечников | 0,1 |
| Глухота врожденная | 0,2 |
| Слепота, рецессивные формы | 0,2 |
| Умственная отсталость неспецифическая | 0,5 |
| Болезнь Тея — Сакса | 0,04 |
| Мукополисахаридоз, тип I | 002 |
| Мукополисахаридоз, тип II | 0,01 |
| Метахроматическая лейкодистрофия | 0,02 |
| Галактоземия | 0,02 |
| Гомоцистинурия | 0,01 |
| Цистинурия | 0,06 |
| Цистиноз | 0,01 |
| Синдром Синта — Ленли — Опитца | 0,01 |
Как обнаруживают генные заболевания?
Для установления наследственной природы заболевания используется клинико-генеалогический метод (генеалогия — родословная). Один из первых этапов изучения генетики наследственного заболевания — выявление больного и составление его родословной. При составлении родословных используются определенные символы (рис. 13). (Далее этими символами будем пользоваться при изложении графического материала.) Ученого-генетика интересует, имел ли кто-либо из родственников больного данное заболевание или оно появилось впервые? Такая постановка вопроса — не праздное любопытство. Ученому необходимо выяснить, что проявилось у больного — новая мутация или старый ген? В зависимости от того или другого прогнозируется риск заболевания потомства не только у пораженного члена семьи, но и у его родственников. Если установлена мутация, то генетик пытается выявить факторы, обусловливающие ее возникновение, для профилактики появления данного заболевания у других членов семьи. Однако что же проявляется чаще при наследственном заболевании — новая мутация или старый ген? Академик АМН СССР Н. П. Бочков писал по этому поводу: «Чем ниже уровень здравоохранения, тем большая доля из общего числа наследственных больных будет определяться новыми мутациями» [1983, с. 55]. Рис. 13. Символы, используемые в генетике человека.
Рис. 13. Символы, используемые в генетике человека.
Установлено, что наследственные заболевания могут иметь аутосомно-доминантный, аутосомно-рецессивный и сцепленный с полом типы наследования. Рассмотрим их подробнее.
Аутосомно-доминантные наследования
При изучении родословных, составленных для некоторых заболеваний, можно установить, что болезнь передается от одного из родителей к детям на протяжении нескольких поколений. Одним из первых наследственных заболеваний человека, описанных в литературе, были брахидактилия (короткие пальцы), синдактилия (сросшиеся пальцы) и полидактилия (добавочные пальцы) (рис. 14—16). Эти признаки имеют аутосомно-доминантный характер наследования. Рис. 14. Брахидактилия у человека.
Рис. 14. Брахидактилия у человека.
 Рис. 15. Родословная больного с синдактилией.
Рис. 15. Родословная больного с синдактилией.
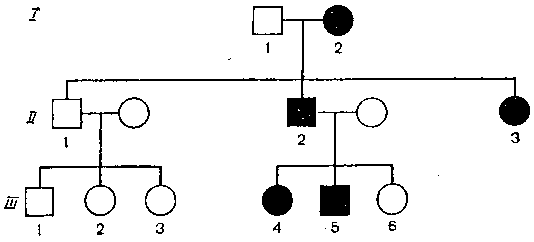 Рис. 16. Родословная больного с полидактилией.
Рис. 16. Родословная больного с полидактилией.
Проследим родословную, в которой полидактилия наблюдалась в трех поколениях (рис. 16). В первом поколении у матери были дополнительные пальцы. Первый ее сын (II-1) не получил аномального гена, и его дети не страдали этим заболеванием. Второй же сын (II-2) и его двое детей (III-4 и III-5) имели дополнительные пальцы. Брахидактилию, синдактилию и полидактилию можно обнаружить уже при рождении ребенка по соответствующему изменению фаланг пальцев рук и ног. Отклонения от нормы в данном случае не угрожают жизни человека, не делают его неполноценным членом общества. Однако существует ряд других аутосомно-доминантных признаков, характеризующихся болезненными проявлениями или даже летальным исходом. Очень тяжелой болезнью является хорея Гантингтона, которая неминуемо вызывает смерть носителя ее гена. Эта болезнь поражает людей после 30 лет, то есть когда у больного, как правило, есть дети. Необходимо отметить, что в нашей стране хорея Гантингтона пока не описана. На рис. 17, 18 показан внешний вид страдающих такими наследственными заболеваниями, как ахондроплазия и микроцифалия.
Для многих известных аутосомно-доминантных генов характерна разная степень их выражения. Это явление открыто генетиком Н. В. Тимофеевым-Ресовским и получило название экспрессивности (степень выражения) генов. С именем Тимофеева-Ресовского связано и другое открытие — явления пенетрантности (частота проявления) генов. С этих позиций вероятность наследственного заболевания определяется степенью выражения и частотой проявления летальных исходов по данному гену (напр., при 100%-й пенетрантности эффект гена проявляется у всех особей, его имеющих, при 50%-й — только у половины). И появляется оптимистический прогноз — не каждый носитель аутосомно-доминантных генов обязательно должен заболеть. Подтверждением этого является следующий пример. Экспрессивностью и пенетрантностью генов можно объяснить передачу по наследству аутосомно-доминантных генов, вызывающих у части обладателей их тяжелейшие заболевания (см. рис. 17, 18).
 Рис. 17. Доминантная карликовость у человека (ахондроплазия).
Рис. 17. Доминантная карликовость у человека (ахондроплазия).
 Рис. 18. Проявление доминантной мутации микроцефалии, связанной со слабоумием.
Рис. 18. Проявление доминантной мутации микроцефалии, связанной со слабоумием.
М. В. Волковым и Е. М. Меерсон в 1973 году описано наследование тяжелого аутосомно-доминантного заболевания — несовершенный остеогенез — в одной семье. Болезнь проявилась у ребенка. Его родители не были поражены таким заболеванием. Однако после детального рентгеновского обследования родственников (выделено восклицательным знаком) стало ясно, что летальный ген, обусловливающий болезнь, передается от отца (рис. 19), у которого, в отличие от сына, получившего наследственный дефект от своих предков, это тяжелейшее заболевание не проявилось. Такое поколение, обладающее нереализованным летальным геном, называется проскакивающим.
 Рис. 19. Родословная больного с несовершенным остеогенезом.
Рис. 19. Родословная больного с несовершенным остеогенезом.
Аутосомно-рецессивные заболевания
При аутосомно-рецессивных заболеваниях у здоровых родителей некоторые дети оказываются больными. Это связано с тем, что оба родителя являются носителями аномального аутосомно-рецессивного гена. Обычно этот ген обозначается буквой «а». С учетом последнего генотип больного по данному гену можно записать как аа, а его родителей (каждого) — как Аа: Как видно из схемы, в потомстве двух гетерозигот могут быть как здоровые, так и больные дети. Среди здоровых также встречаются гетерозиготы. Чаще всего в семье бывает два ребенка. Семьи с двумя детьми от родителей-гетерозигот можно разделить на три группы: 1 — нет больных детей (такие случаи редки); 2 — болен один ребенок; 3 — больны оба ребенка.
Однако у родителей-гетерозигот, так же как и у всех, могут рождаться близнецы. Если последние однояйцевые, то оба будут либо больными (гомозиготы), либо здоровыми(гетерозиготы или не имеющие вообще аномального гена). В случае же разнояйцевых близнецов один из них может быть здоровым, а другой — больным.
Как видно из схемы, в потомстве двух гетерозигот могут быть как здоровые, так и больные дети. Среди здоровых также встречаются гетерозиготы. Чаще всего в семье бывает два ребенка. Семьи с двумя детьми от родителей-гетерозигот можно разделить на три группы: 1 — нет больных детей (такие случаи редки); 2 — болен один ребенок; 3 — больны оба ребенка.
Однако у родителей-гетерозигот, так же как и у всех, могут рождаться близнецы. Если последние однояйцевые, то оба будут либо больными (гомозиготы), либо здоровыми(гетерозиготы или не имеющие вообще аномального гена). В случае же разнояйцевых близнецов один из них может быть здоровым, а другой — больным.
Таблица 8. Частота встречаемости фенилкетонурии и галактоземии среди новорожденных в разных странах
| Страна | Число обследованных новорожденных | Частота встречаемости фенилкетонурии |
|---|---|---|
| Польша | 894 891 | 1 : 7 782 |
| Чехословакия | 132 392 | 1 : 6 618 |
| Австрия | 666 383 | 1 : 12 340 |
| Швейцария | 699 089 | 1 : 16 644 |
| Франция | 1 882 734 | 1 : 13 715 |
| ФРГ | 359 875 | 1 : 10 935 |
| Дания | 285 535 | 1 : 11 898 |
| Швеция | 907 746 | 1 : 43 226 |
| Финляндия | 71 111 | 1 : 71 111 |
| Англия | 112 362 | 1 : 10 215 |
| Ирландия | ||
| западные области | 825 935 | 1 : 5 343 |
| восточные // | 206 011 | 1 : 7 924 |
| США | 1408 425 | 1 : 13 630 |
| Канада | 277 769 | 1 : 39 681 |
| Новая Зеландия | 381 536 | 1 : 18 168 |
| Австралия | 353 458 | 1 : 9 818 |
| Япония | 210 851 | 1 : 210 851 |
| Израиль | ||
| евреи ашкенази | 180 000 | 1 : 180 000 |
| другие евреи, арабы | 320 000 | 1 : 8 648 |
| Страна | Число обследованных новорожденных | Частота встречаемости галактоземии |
| Польша | 307 947 | 1 : 12 317 |
| Чехословакия | 132 392 | 1 : 44 130 |
| Австрия | 664 966 | 1 : 39 116 |
| Швейцария | 520 456 | 1 : 65 057 |
| Бельгия | 106 511 | 1 : 10 651 |
| ФРГ | ||
| северные области | 119 024 | 1 : 29 756 |
| западные // | 300 355 | 1 : 42 883 |
| Швеция | 907 746 | 1 : 49 000 |
| Ирландия | 144 842 | 1 : 482 188 |
| США | 732 911 | 1 : 104 701 |
| Канада | 148 872 | 1 : 148 872 |
| Новая Зеландия | 292 626 | 1 : 32 514 |
 Рис. 20. Локализация генетического блока при фенилкетонурии. а, б — путь метаболизма фенилаланина соответственно у больных и здоровых.
Рис. 20. Локализация генетического блока при фенилкетонурии. а, б — путь метаболизма фенилаланина соответственно у больных и здоровых.
Рассмотрим случай: жених здоров, у него нет слабоумия, однако гены в обеих матрицах остались гомозиготными — аа. Он собирается организовать семью. Если окажется, что у невесты нет ни одного гена, обусловливающего появление фенилкетонурии, то все их дети будут гетерозиготы (Аа) — то есть здоровы. Если же он полюбит девушку, гетерозиготную по гену фенилкетонурии (Аа), то в 50 % случаев можно ожидать рождения больного ребенка. Теперь рассмотрим случай, когда «вылеченная» девушка стала невестой и собирается выйти замуж. Будет ли картина аналогичной предыдущей? Здесь все оказалось сложнее. «Вылеченная» девушка собирается стать матерью. Генетический дефект же у нее остался. Аминокислота фенилаланин в повышенной концентрации уже не может повлиять на ее мозговые клетки, однако будет губительно действовать на мозговое вещество ребенка, находящегося в утробном развитии. Каролин и Стивенсон в 1969 году описали течение беременности у двух женщин, больных фенилкетонурией. 26 беременностей у пациенток закончились 16 спонтанными абортами в первые три месяца и 10 живорожденными детьми. Уровень фенилаланина в крови детей был выше, чем в крови матери. Живорожденные дети имели следующие пороки развития: микроцефалию, врожденные пороки сердца, умственную отсталость и др. По логике, фенилаланин должен быть в повышенных концентрациях и у женщин, гетерозиготных из-за недостаточной активности фермента. Медики-генетики стали искать женщин-гетерозигот по фенилкетонурии среди населения и наблюдать за течением беременностей у них. Оказалось, что только у 6 из 46 таких женщин беременность и роды протекали нормально. Из сказанного ясно, что гетерозиготное носительство гена фенилкетонурии со стороны матери, не говоря уже о гомозиготном (больная мать),— чрезвычайно неблагоприятное явление для беременности и родов. Несколько слов о гениальности и таланте. Гениальность, как и талант человека, является результатом взаимодействия генотипа (со всем комплексом его врожденных данных) и окружающей среды. Соотношение этих двух слагаемых для каждого конкретного признака (свойства) разное, однако оба они в развитии вышеуказанных качеств необходимы. При благоприятной среде и отсутствии необходимого комплекса генов эти качества не разовьются. И, напротив, при предрасположенности к тому или иному дарованию, не подкрепленной необходимой социальной средой, результат будет аналогичным. Однако, опираясь на данные современной науки, можно констатировать, что в гениальности ученого, поэта, писателя, художника и т. д. вклад генотипа преобладает. Но здесь нет места для пессимизма. Каждый здоровый человек талантлив по-своему и представляет для общества громадную ценность, и задача общества (и семьи) в том, чтобы это качество выявить и развить. Современная генетика доказала, что человек является продуктом биосоциальным, а не «чисто социальным», как утверждали классики, и что его биологическая компонента составляет около 70%, а такие властные инстинкты, как размножение, самосохранение, забота о потомстве и т. д.,— все 100 %.
Гены, сцепленные с половыми хромосомами
К настоящему времени известно более 200 генов (доминантных и рецессивных), сцепленных с Х-хромосомой, то есть локализованных в ней. Многие из них обусловливают появление наследственных болезней (гемофилия, мышечная дистрофия и др.) (рис. 21). У женщин аномальный ген может находиться в одной (гетерозигота) или обеих (гомозигота) Х-хромосомах, у мужчин — только в одной Х-хромосоме. Наиболее широко распространено заболевание, обусловленное геном, сцепленным с Х-хромосомой и имеющим рецессивный характер наследования,— гемофилия (несвертываемость крови) (рис. 22). В данном случае ген имеет полулетальное значение, что, как правило, приводит к гибели ребенка в раннем возрасте. Долгое время считалось, что этим заболеванием страдают только мужчины. Однако оно встречается и у женщин, хотя и крайне редко, и обусловлено наличием аномальных генов в обеих Х-хромосомах.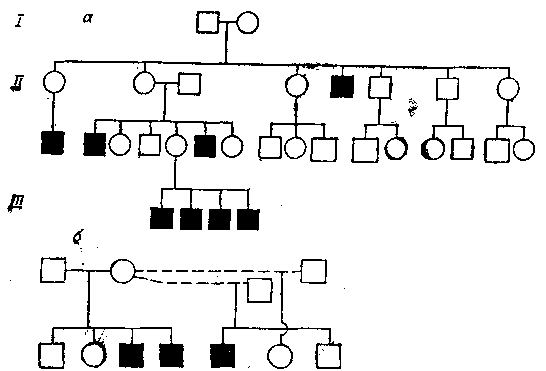 Рис. 21. Наследование рецессивного гена мышечной дистрофии Дюшена, локализованного в Х-хромосоме.
а — родословная трех поколений с мышечной дистрофией только среди мужчин; б — родословная трех больных мальчиков, имеющих одну мать, но двух отцов.
Рис. 21. Наследование рецессивного гена мышечной дистрофии Дюшена, локализованного в Х-хромосоме.
а — родословная трех поколений с мышечной дистрофией только среди мужчин; б — родословная трех больных мальчиков, имеющих одну мать, но двух отцов.
Рассмотрим случай — мать здорова, отец болен гемофилией (рис. 23, а). Их дочери будут здоровыми, так как получат лишь одну Х-хромосому с аномальным геном (от отца). Сыновья также будут здоровы, так как им от отца перейдет Y-хромосома (без аномального гена). При больной матери и здоровом отце, ситуация иная (рис. 23, б). В этом случае их дочери будут всегда здоровы, сыновья же могут быть и больными, и здоровыми в зависимости от того, перейдет или не перейдет к ним от матери аномальный ген. Третий случай — больные и мать, и отец (рис. 23, в). Здесь страдают болезнью и сыновья, и дочери. Первые обязательно получат аномальную Х-хромосому от матери, а вторым обязательно перейдут по одной аномальной Х-хромосоме от отца и матери, а гомозиготность обусловит болезнь. Как упоминалось выше, этот случай встречается крайне редко,
 Рис. 22. Генеалогическое древо царствовавших семей в Европе, иллюстрирующее наследование гена с гемофилией, локализованного в X-хромосоме. Усл. обозн., см. на рис. 13.
Рис. 22. Генеалогическое древо царствовавших семей в Европе, иллюстрирующее наследование гена с гемофилией, локализованного в X-хромосоме. Усл. обозн., см. на рис. 13.
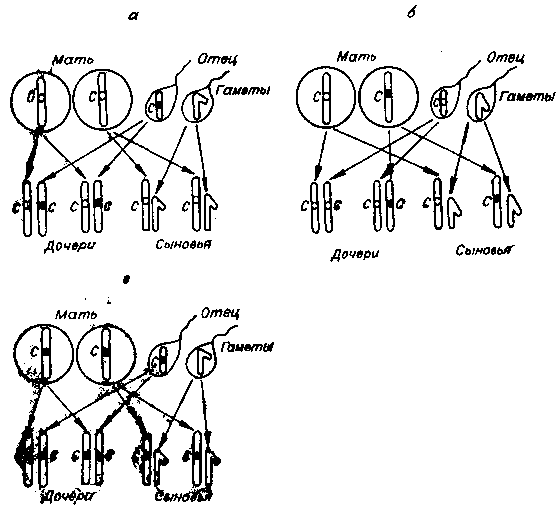 Рис. 23. Наследование гена гемофилии, а — мать здорова, отец болен гемофилией; б — отец здоров, мать больна гемофилией; в — больные и мать, и отец.
Рис. 23. Наследование гена гемофилии, а — мать здорова, отец болен гемофилией; б — отец здоров, мать больна гемофилией; в — больные и мать, и отец.
Болезнь мышечной дистрофии Дюшена поражает мальчиков в возрасте от 2 до 6 лет, начинаясь с атрофии мышц грудной клетки и поясничной области, позже охватывающей мускулатуру конечностей. Это заболевание известно давно, однако только в последний годы ученые выяснили механизм действия аномального гена. Болезнь имеет рецессивный тип наследования и ген локализован в Х-хромосоме. Был обнаружен белок, названный дистрофином, на долю которого приходится 0,002 % общего количества белка в нормальных скелетных мышцах. Отсутствие дистрофина у больных мышечной дистрофией ученые связывают с дефектом гена. Несмотря на малые количества этот белок является важным компонентом мышечной ткани и отсутствие его в последней приводит к распаду мышц. Считается, что дистрофия играет основную роль в контроле над внутренней средой мышечной клетки, предотвращая бесконтрольное повышение концентрации кальция в клетке. При нарушении кальциевого равновесия активируется фермент фосфолипаза А, растворяющий мышечные волокна. Мышцы скелета, пытаясь восстановить это нарушение, подвергаются фиброзу (процессу затвердевания), который и нарушает мышечную функцию. Явление фиброза пока изучено недостаточно. Имеется гипотеза, что даже при отсутствии дистрофина мышечные клетки могли бы восстанавливаться, но вторичная патология — фиброз — препятствует этому. Последнее подтверждается тем, что в организме известны мышцы, в которых дистрофии отсутствует, однако дистрофии в них не наблюдается. В этих случаях, очевидно, мышцы не подвергаются фиброзу.
Глава 4. Наследственность, болезни
Болезни с наследственным предрасположением
Выше мы рассмотрели группу наследственных заболеваний, причиной которых является наличие в генотипе человека мутантных генов, обусловливающих данные дефекты. Однако эта группа отклонений невелика и составляет примерно 6—8 % от общего числа болезней, которыми страдает человек. Наиболее часто встречаемые болезни — с наследственным предрасположением (90—92 %). Это сахарный диабет, сердечно-сосудистые и аллергические заболевания, атеросклероз, язва желудка и двенадцатиперстной кишки, ревматизм, шизофрения, врожденные пороки развития и др. При перечисленных болезнях из поколения в поколение передается предрасположенность к определенному заболеванию. Наследственные заболевания — одна из основных причин смерти новорожденных. Наиболее часты врожденные аномалии опорно-двигательного аппарата, нервной и сердечно-сосудистой систем, выявляемые в первый год жизни ребенка (в США в среднем у 3 % детей). До 14-летнего возраста врожденные аномалии в разных странах занимают второе или третье место среди причин детской смертности. Проявление конкретного заболевания зависит не только от генотипа, но и от факторов внешней среды, в которой находится человек. К последним относятся место проживания — город или село, задымленность и загазованность воздуха, шум; характер питания — частота приема пищи, ее разнообразие, качество и количество; характер деятельности человека — напряженная (легкая) умственная или физическая, приносящая удовлетворение, или вызывающая раздражение работа. Одни из этих факторов могут способствовать появлению болезней с наследственным предрасположением, другие, наоборот, играют по отношению к ним профилактическую роль. Рассматриваемые болезни — полигенные, то есть обусловленные несколькими генами, заболевания. Эти гены могут проявиться и в гетерозиготном состоянии. Изучение болезней с предрасположением — одна из актуальных задач современной генетики. Доказано, например, что ишемическая болезнь сердца встречается в 5 раз чаще, а сахарный диабет — в 10 раз чаще среди родственников больного, чем в популяции в целом. Пока нет полных сведений о характере генетического влияния на все болезни с наследственным предрасположением. Однако известно, что, например, сахарный диабет — заболевание генетически гетерогенно, то есть разные дефекты различных генов могут обусловливать клиническую картину данной болезни. Выделяют инсулинзависимый, неинсулинзависимый и другие виды сахарного диабета. Установлено, что инсулинзависимый диабет характеризуется аутосомно-рецессивным типом наследования, однако он проявляется только у одной из двух гомозигот. Частота встречаемости этого вида диабета в популяции весьма велика — 1:7,6. Неинсулинзависимый сахарный диабет характеризуется аутосомно-доминантным типом наследования, причем по наследству передается лишь предрасположенность к нему. Рассмотрим некоторые болезни с наследственным предрасположением более подробно.Сахарный диабет
Сахарный диабет — широко распространенное заболевание. По данным Всемирной организации здравоохранения (ВОЗ), в настоящее время сахарным диабетом болеют более 30 миллионов человек (около 4,5 % населения промышленно развитых стран) и, более того, наблюдается тенденция к увеличению этого заболевания. При сахарном диабете в большей мере чем при других заболеваниях сохранение здоровья зависит от образа жизни человека. Больной и его родственники должны иметь полную осведомленность о своей болезни, остроте течения, возможных осложнениях, лечении, режиме повседневной жизни. Исследования последних лет показали, что сахарный диабет — аутоиммунное заболевание, то есть отличается аутоиммунной реакцией на собственные бета-клетки, которые вырабатывают инсулин. Аутоантитела удается обнаружить у лиц, предрасположенных к диабету, еще за 7 лет до появления симптомов болезни. Таким образом, найден ранний маркер диабета. Если вовремя будет поставлен диагноз, то применением циклоспорина, останавливающего аутоиммунную реакцию в организме, можно не допустить возникновения болезни. Лечение циклоспорином позволило прекратить инъекции инсулина в одном случае у 25 % больных (из 188), а в другом — у 67 % (из 40). Успех лечения больных сахарным диабетом во многом зависит от времени начала лечения. По мнению американских ученых, болезнь можно предотвратить, если ее лечить с детского возраста. В 1988 году на съезде, организованном американской ассоциацией по изучению сахарного диабета, были доложены результаты, полученные в последние годы. К этому времени было установлено, что в развитии болезни главное значение имеет не генетический, а внешний фактор, определяемый условиями жизни и характером жизнедеятельности человека. Ученые считают, что не менее 60 % заболеваний сахарным диабетом, включая инсулинзависимый, можно предотвратить. Американские генетики пришли к выводу, что причиной широкого распространения в последние годы сахарного диабета среди детей в возрасте до 15 лет в северо-восточной части США явились пагубные изменения в среде обитания человека в этом регионе. «Вспышки» болезни имели место и в других странах. Так, в Польше в 1981—1984 годах заболеваемость диабетом среди детей некоторых районов страны возросла в 2 раза. Причина этого не была установлена, но ученые предполагают, что болезнь имела вирусное происхождение. Однако и вирусы, и другие агенты стимулируют развитие болезни там, где она была предопределена генетически.Сердечно-сосудистые заболевания
Существует мнение, что наследственность является одной из главных (если не самой главной) причин высокой смертности при болезнях сердца и сосудов, нарушениях обмена веществ и других болезнях крови. Последнее подтверждается тем, что среди братьев и сестер люди с врожденными аномалиями сердца встречаются в 4—6 раз чаще, чем в популяции в целом. Частота врожденных аномалий сердца составляет 6:1000. В литературе [Петросян и др., 1971] описаны семейные случаи врожденных аномалий сердца, наследующихся по аутосомно-доминантному типу. Наследственный характер доказан и для синдромов, характеризующихся сочетанием врожденных пороков сердца с аномалиями костной системы (синдромы Хольта — Омара, Льюиса, стеноз аорты с гиперкальцемией, врожденный порок сердца с синдактелией и др.). Установлено, что у больных с хромосомными отклонениями врожденные аномалии сердца, обусловленные нарушением числа хромосом, составляют примерно 1:200. В связи с врожденными сердечно-сосудистыми заболеваниями наиболее интересен синдром Шерешевского — Тернера (половые хромосомы Х0). Классический тип этого синдрома сопровождается каортацией аорты, нередко наблюдается и стеноз легочной артерии. Цитогенетическими методами показано, что существует много видов синдрома Шерешевского — Тернера, связанных с мозаицизмом хромосом. В женском организме содержится разное количество клеток с нормальным набором половых хромосом (XX), что определяет большую вариацию в клинической картине этого заболевания — от явно выраженного синдрома до почти нормального состояния, хотя в последнем случае женщины часто имеют низкий рост, а в период полового созревания у них выявляются и эндокринные расстройства. В. П. Эфроимсон [1965] считает необходимым проводить тщательный анализ хромосом у всех женщин низкого роста с эндокринными расстройствами. Пороки сердца наблюдаются и при аномалиях аутосом. Трисомии D и Е в 100% случаев сопровождаются множественными пороками сердца и сосудов и особенно часто дефектом перегородок сердца. Установлено, что до 60 % больных с аномалиями в 21-й хромосоме (болезнь Дауна) имеют врожденный порок сердца. В 1971 году в 57 % таких больных выявлены структурные нарушения и в 16-й хромосоме. Более тщательные исследования японских ученых 22 детей с врожденными пороками сердца у 9 из них выявили аномалии в 16-й хромосоме. При использовании методики дифференциальной окраски хромосом выявлено, что характер и расположение полос специфичны, что дает возможность для каждой пары различить (и выявить) хромосомы в пределах каждой группы. В 1973 году начаты работы по изучению связи врожденных сердечно-сосудистых заболеваний о возрастом родителей. Оказалось, что частота аномалий увеличивается с возрастом матери и порядковым номером родов. Кроме того, для каждого типа пороков сердца наблюдается увеличение возраста родителей.Аллергические заболевания
За последние годы почти во всех странах мира отмечен значительный рост аллергических заболеваний. Статистика показывает, что на долю этих заболеваний приходится более 10%. Причинами их роста являются применение прививок, все возрастающий контакт людей в быту и на производстве с химическими веществами, в том числе рост потребления лекарственных препаратов. Впервые термин «аллергия» введен в медицину в 1906 году. Он происходит из двух слов: «аллос» — другой, иной и «ергия» — действие, реакция. Иными словами, аллергия — это иная, необычная, реакция организма на агенты различного происхождения — аллергены. Аллергены, в свою очередь — это вещества, способные проникать в организм и вызывать изменения в его обменных функциях. Все аллергены делятся на экзо- и эндоаллергены (или аутоаллергены). Первые попадают в организм извне, вторые образуются в самом организме. Каждый аллерген может содержать от одного до нескольких антигенов А (антигены — чужеродные органические соединения преимущественно с большой молекулярной массой). Как установлено, многие простые химические вещества (бром, иод, хлор и др.) и сложные соединения небелковой природы при попадании в организм могут вызывать аллергию. Однако эти вещества становятся антигенами (аллергенами) только после соединения с белками тканей организма. В основе аллергических заболеваний лежат иммунологические механизмы, то есть отклонения затрагивают систему иммунитета. Термин «иммунитет» многим уже знаком в связи с распространением в последние годы вирусного заболевания, поражающего имунную систему и пока не поддающегося лечению,— синдрома приобретенного иммунодефицита (СПИД), называемого вирусом иммунодефицита человека (ВИЧ). К числу аллергических заболеваний относятся бронхиальная астма, поллиноз (аллергия к пыльце растений), аллергический контактный дерматит, лекарственные аллергии и др. У человека по наследству передается предрасположенность к аллергии вообще, но не к ее конкретной форме. Для выяснения механизмов, вызывающих какое-либо аллергическое заболевание, а также для отработки способов его лечения предприняты попытки обнаружить или вызвать такое же заболевание у животных. Группе американских ученых удалось найти у мышей два гена, обусловливающих у них развитие астмы. Эти гены могут вызвать гиперчувствительность бронхов (повышенную бронхиальную реактивность), что является основным фактором в развитии астмы. Изучение семей, в которых астма является наследственной болезнью, показало повышенную бронхиальную активность даже у тех членов семьи, которые не страдают астмой. А это значит, что у них имеется генетический фактор. Один из генов, обнаруженный у мышей, наследуется как рецессивный фактор, и для того, чтобы развилась повышенная бронхиальная реактивность, потомство должно наследовать дефектный ген от обоих родителей. У человека пока таких генов не обнаружено. Группе ученых удалось выделить из кормового растения райграс ген, который вырабатывает переносимый пыльцой белок, вызывающий сенную лихорадку (поллиноз). Вероятно, следующим этапом исследований будет создание с помощью этого гена синтетических пептидов (белков) с целью использования их в качестве диагностических и лечебных средств. Конечная цель этих работ — создание вакцин против сенной лихорадки и астмы. Лекарственная аллергия. К настоящему времени количество лекарственных препаратов возросло до 400 тыс. наименований. (В этой связи кстати напомнить, что безвредных для организма препаратов нет.) Лекарственная аллергия — один из побочных эффектов действия лекарств. Поэтому для профилактики лекарственной аллергии рекомендуется не применять лекарственные препараты без назначения врача, и своевременно сообщать врачу о препаратах, вызывающих у вас аллергию. При наличии воспалительных процессов на коже вокруг места инъекций необходимо обратиться к врачу, так как указанный признак может служить первым сигналом к отмене применения препарата. Пищевая аллергия. Развитие пищевой аллергии обусловлено повышенной проницаемостью кишечнопеченочного барьера для пищевых антигенов, что приводит к всасыванию не полностью расщепленного (гидролизованного) белка пищевых продуктов. Ведущий аллерголог нашей страны В. А. Адо с сотрудниками [1982] считают, что причиной широкого распространения пищевой аллергии часто является избыток потребляемых продуктов, а также применение в пищевой промышленности красителей и консервантов, а в сельском хозяйстве — химических удобрений и ядохимикатов. До последнего времени пищевая аллергия привлекала внимание педиатров, так как в детском возрасте отмечается повышенная проницаемость кишечно-печеночного барьера для пищевых антигенов. У взрослых пищевая аллергия протекает часто в замаскированной форме, имитируя хронические заболевания разных органов. Пищевую аллергию могут вызывать любые продукты питания, но наиболее часто — самые распространенные в данной местности. При данном заболевании могут наблюдаться признаки поражения пищевого тракта (полости рта, пищевода, желудка, кишечника и толстой кишки). Аллергические симптомы могут появиться и вне пищеварительной системы — повышение температуры тела, крапивница, различные отеки, головная боль, озноб, лихорадка и др.Язвенная болезнь
Известно, что возникновение и характер течения язвенной болезни во многом зависят от того, насколько внимательно человек относится к своему здоровью, соблюдает режим питания, воздерживается от вредных привычек, таких как курение, прием алкоголя и др. Несоблюдение этих установок, и в первую очередь нерегулярное и некачественное питание, часто является причиной возникновения язвенной болезни, особенно в молодом возрасте. Среди врачей бытует даже такой термин, как «студенческая язва», то есть случаи развития язвенной болезни среди учащихся, питающихся нерегулярно, всухомятку, от случая к случаю. Однако при одинаковых сопутствующих причинах язвенное заболевание развивается не у всех, а только у незначительной части лиц. Следовательно, существуют люди, подверженные этой болезни в большей или меньшей степени. В 1936 году советские ученые А. Е. Левин и Б. А. Качур, проанализировав большой материал, впервые обратили внимание на то, что в семьях больных язвенной болезнью последняя встречается значительно чаще, чем в семьях здоровых людей. Этот факт, неоднократно подтвержденный в последующих исследованиях и получивший название «семейное накопление» язвенной болезни, может быть связан с определенной генетической общностью родственников, в результате чего некоторые члены семей больных этой болезнью наследуют генетические факторы, предрасполагающие к развитию заболевания. Казалось бы, не исключено и то, что «семейное накопление» может также определяться характерными внутрисемейными факторами, способствующими развитию язвенной болезни: традициями быта, питания, вредными привычками и т. д. Ученые не отвергают такой возможности. Однако язвенная болезнь гораздо чаще развивается у лиц, у которых уже с юношеского (а иногда и с детского) возраста отмечаются определенные отклонения в функционировании желудочно-кишечного тракта (в частности, повышенное выделение желудочного сока). Последнее может быть связано с врожденным увеличенным количеством клеток в слизистой оболочке желудка, которые выделяют соляную кислоту и другие компоненты желудочного сока. Существенна и такая закономерность: в семьях, где отмечается несколько случаев язвенной болезни, последняя протекает, как правило, более тяжело, а родственники чаще всего страдают одной и той же формой заболевания — язвенной болезнью либо двенадцатиперстной кишки, либо желудка. По результатам исследований, выполненных в Институте медицинской генетики АМН СССР, доля наследственных факторов в развитии язвенной болезни составляет около 60 %, а социальных факторов — около 40 %. Необходимо отметить, что учеными не подтверждено существовавшее ранее мнение о том, что практически во всех случаях имеет место «прямая» передача язвенной болезни из поколения в поколение, как это отмечается при некоторых других генетических заболеваниях. Все вышесказанное относится к так называемому формальному этапу в истории генетики язвенной болезни. Новый этап, характеризующийся поиском конкретных генетических факторов, предрасполагающих к данному заболеванию, начался с неожиданного открытия. В 1954 году было обнаружено, что среди больных язвенной болезнью по сравнению со здоровыми людьми больше лиц с первой группой крови. Это явление, получившее название ассоциации (связи) язвенной болезни с первой группой крови, сразу привлекло внимание ученых разных стран. Последовало большое число исследований, направленных на выявление подобных связей язвенной болезни с другими генетическими признаками. Многие из этих поисков увенчались успехом. Так, в 1956 году была найдена связь язвенной болезни с генетическим признаком «несекретор» — отсутствием выделения в слюну и желудочный сок определенных антигенов. В дальнейшем ученые обнаружили корреляции язвенной болезни с генетическими признаками, определяющими группу и биохимические свойства крови. В чем же биологический смысл выявленных корреляций язвенной болезни с генетическими факторами? На этот счет было предложено много гипотез, однако ни одна из них так и не дала полного объяснения данному удивительному феномену. Тем не менее стало ясно, что наследственность при язвенной болезни, действительно, играет определенную роль. Ученые рассуждали так: если какой-то генетический признак чаще встречается среди больных язвенной болезнью по сравнению со здоровыми, но наблюдается не у всех больных, а только у части (пусть иногда и незначительной), следовательно, наличие этого признака еще не обрекает человека на заболевание, а лишь предрасполагает к нему, то есть повышает риск развития язвенной болезни по сравнению с теми людьми, кто этим признаком не обладает. Так было сформулировано и введено в медицинскую практику понятие «фактор риска», наличие которого у человека повышает вероятность развития у него той или иной болезни. Полученные данные о «факторах риска» имеют большое значение для прогнозирования заболеваемости. Специальными расчетами показано, что у лиц с II—IV группами крови по сравнению с обладателями I группы фактор риска возрастает на 10%, а наличие у человека признака «несекретор» повышает риск заболевания на 60%. При сочетании этих двух генетических признаков «фактор риска» возрастает уже в 2,25 раза. Данные о степени риска можно получить и по наличию других признаков, связанных с язвенной болезнью. Практическая ценность такого подхода заключается в возможности профилактического обследования большого количества здоровых людей для выявления среди них тех, у кого, по данным такого обследования, риск развития язвенной болезни окажется повышенным. Этих лиц, относящихся к так называемой группе риска по язвенной болезни, уведомляют о результатах обследования для повышения их внимания к режиму, организации питания и другим факторам и ставят на специальный диспансерный учет для проведения профилактических осмотров. Первичная профилактика заболевания является основной задачей здравоохранения. Такая же цель стоит и перед медицинской генетикой, достижения которой уже сегодня позволяют подойти к решению этой проблемы. Так, в Институте медицинской генетики АМН СССР создана программа по прогнозированию развития язвенной болезни, с помощью которой появляется возможность отбора (например, при профосмотрах) лиц с повышенным риском развития заболевания. Итак, в настоящее время можно с уверенностью утверждать, что успехи медицинской генетики опровергают бытовавшее ранее мнение о якобы фатальной роли наследственности, о том, что человек, имеющий аномальные (или только предрасполагающие к заболеванию) гены, бессилен перед болезнями. Доказано неоспоримо, что генетические факторы риска далеко не в каждом случае вызывают заболевание. И здесь с целью профилактики заболевания большое значение для человека имеют правильный режим питания, отказ от вредных привычек, соблюдение режима труда и отдыха, физическое закаливание. Многочисленные примеры свидетельствуют, что при условии правильного подхода человека к охране собственного здоровья даже напряженный физический или умственный труд не приводит к развитию заболевания.Питание и болезни
То, что питание является одним из важнейших условий для сохранения здоровья, доказывать, видимо, никому не надо. Оно выполняет две задачи: 1 — снабжения организма энергетическим и строительным материалом для постоянного обновления клеток; 2 — выработки гормонов, ферментов и плазмы крови. Питание является правильным в том случае, если по количеству и составу оно удовлетворяет эти потребности организма. А последние определяются тем, что на 1 кг массы человека в сутки необходим 1 г сбалансированного белка. Сбалансированность, в свою очередь, характеризуется наличием незаменимых аминокислот в потребляемом белке. Содержание белка, например, в разных сортах мяса колеблется в пределах 15—20 %. Чтобы человеку массой 80 кг чувствовать себя физиологически здоровым, ему необходимо потреблять в сутки 400 г мяса (80 * 5). К этому, конечно, необходимо добавить около 1 кг овощей и фруктов. О том, много ли Вы едите или мало, можно судить по Вашей массе. Существует несколько формул для расчета оптимальной массы тела. Так, по Броку, масса тела (кг) = рост (см) — 100—8 % (8 % от полученной впереди разности); по Бернгарду, масса тела (кг) = рост (см) * объем груди (см) / 240. Избыток массы, как правило, свидетельствует о неправильном процессе обмена веществ. Однако на последнее может указывать и нормальная масса. Иными словами, для профилактики здоровья предпочтительно иметь массу, немного меньшую нормальной. Необходимо помнить, что каждое переедание вредит здоровью и сокращает жизнь. Лишняя масса вызывает следующие негативные явления: 1 — перегрузку на органы кровообращения из-за необходимости постоянно снабжать большое количество тканей тела; 2 — повышение расхода энергии при движении; 3 — перегрузку органов дыхания из-за повышенной потребности организма в кислороде, что ведет к уменьшению жизненной емкости легких; 4 — смещение диафрагмы кверху жировыми слоями в области живота, обусловливающее высокое давление, склонность к инфаркту, поражение сердечной мышцы, варикозное расширение вен, дыхательную недостаточность и другие негативные проявления; 5 — излишнюю статическую нагрузку на скелетную систему, что повышает предрасположенность к изменениям в суставах конечностей и в позвоночнике. Предрасположенность к ожирению имеет наследственный характер. В семьях, где оба родителя имеют нормальную массу, только у 9 % детей масса превышает нормальную. В семьях же, где один или оба родителя тучны, от 60 до 80 % детей имеют ожирение. Однако известно, что помимо наследственного предрасположения к тучности еще многие факторы провоцируют возникновение ожирения. Такими факторами могут быть переедание, малоподвижный образ жизни, инфекция и интоксикация, поражение центральной нервной системы и др. При этом переедание является причиной ожирения номер один, и 60—80 % больных страдают тучностью именно из-за него [Шурыгин и др., 1980]. Ожирение стимулирует развитие в организме таких заболеваний, как гипертония, атеросклероз, сахарный диабет, цирроз печени, желчно-каменная болезнь, печеночно-каменная болезнь и др. Эти заболевания ведут к сокращению жизни больных в пожилом возрасте на 10—12 лет. Необходимо отметить, что мужчины и женщины имеют разные точки зрения на свою массу. Так, женщины чаще мужчин стремятся похудеть, даже если их масса нормальна. Средством похудения мужчины в основном избирают физические упражнения, а женщины — диету, которая является главным фактором риска в отношении развития недугов, связанных с неправильным питанием. Совершенно необоснованно, когда из диеты исключаются жиры. Установлено, что недостаточное поступление жира с пищей ведет к ослаблению иммунных свойств организма. Более того, в экспериментах с использованием культуры раковых клеток молочной железы мышей показано, что полиненасыщенные кислоты — линолевая, гамма-линолевая и архидониевая — обладают свойством убивать раковые клетки. В опытах на мышах показано, что защитным эффектом обладает и сливочное масло. В развитии ожирения большое значение имеет качественный состав пищевого белка. Отсутствие в последнем оптимальных соотношений незаменимых аминокислот способствует увеличению количества жира в организме, что, в свою очередь, ведет к излишней массе. Неблагоприятно для организма и недоедание. Последнее возникает не только из-за отсутствия пищи, но и вследствие других причин: однообразия пищи, нарушенного пищеварения, искусственного вызывания рвоты после еды, часто из-за «голодных» диет, применения сразу после еды слабительных и мочегонных средств. Недоедание, так же как и переедание, вызывает нарушение функциональной деятельности отдельных органов и организма в целом. Многих, наверное, поразят слова Гиппократа: «Человек рождается здоровым, все болезни приходят к нему через рот с продуктами питания». В компетентности этого врача, а следовательно, и верности его суждения сомневаться не приходится. Поэтому организация правильного питания — для каждого человека одна из главных задач. Однако это особый вопрос и особая тема, выходящая за пределы данной публикации.Повреждение эмбриона в период беременности
Период вынашивания плода — крайне ответственное время для формирования здорового организма потомства. Установлено, что из 4,5 % детей, родившихся неполноценными, 1,0—1,5 % имеют отклонения,вызванные отнюдь не аномалиями в генах. Причина трагедии здесь в том, что мамы в силу различных причин — незнания, низкой культуры, приверженности вредным привычкам — нещадяще относятся в период беременности к будущему ребенку. Эмбрион часто повреждается лекарствами, используемыми матерью без врачебного надзора. Инфекционные заболевания вирусной природы (желтуха-гепатит, вирус Герпеса-зоостер и др.), перенесенные беременной женщиной, довольно часто приводят к рождению ребенка с различными отклонениями. На весь мир прозвучали факты применения в Федеративной Республике Германии непроверенных на наследственность лекарств (например, обезболивающего препарата талидомида), приведшие к рождению около 6 тысяч детей-инвалидов (без рук, ног и с другими физическими недостатками). По требованию общественности эти дети собраны в государственные детские дома инвалидов, где их пытаются научить элементарным навыкам ухода за собой. Нельзя забывать, что в течение девяти месяцев беременности, когда из двух половых клеток возникает и формируется ребенок, происходят тысячи (десятки тысяч) химических реакций, «работают» многие гены, образуются различные ферменты. Появление в это время в организме матери алкоголя, никотина, лекарственных препаратов или вирусных инфекций может привести к повреждению плода и сделать ребенка на всю жизнь больным, а семье, вместо радости, принести трагедию. Возьмем, например, самое, казалось бы, «безобидное», часто употребляемое большинством населения, лекарство — аспирин. Этот препарат сравнительно давно внедрен в медицинскую практику и нашел в ней широкое применение, однако механизм его действия стал известен только в последние годы. Установлено, что аспирин (синоним — ацетилсалициловая кислота) поражает легочную и печеночную ткани у эмбриона до трех месяцев. Степень этого поражения зависит от характера питания матери: на фоне углеводной диеты поражение будет сильным, на фоне высокобелковой — гораздо меньшим. В каждой семье имеется домашняя аптечка, и до вызова врача люди часто применяют лекарства сами. Однако для беременной женщины такая самостоятельность крайне опасна. Решение о том, какое лекарство можно применять в данном случае, может принять только врач, знающий механизмы действия различных препаратов. Только врач посоветует и правильную диету. Опасны для будущих новорожденных и вирусные заболевания, упомянутые выше. Из 100 женщин, подверженных в первые три месяца беременности действию вируса желтухи-гепатита, 3—5 % рожают детей, пораженных микроцефалией (маленькая голова). Внешне микроцефалики имеют признаки полных идиотов и не поддаются в этом плане какой-либо коррекции. Другой, казалось бы, безобидный вирус Герпеса-зоостер, поражая беременных женщин, в 3—4 % случаев вызывает повреждение плода, делает будущего человека инвалидом. В период беременности будущая мать должна думать о здоровье не только своего ребенка, но и будущих внуков. Напомним, что первое редукционное деление в мейозе (образование половых клеток) происходит у человека в то время, когда он еще находится в утробе матери. Питание будущих матерей должно быть разнообразным, содержать необходимые количества белков, витаминов, минеральных солей. При недостатке последних возможны различные нарушения в развитии эмбриона. Питаясь в период формирования ребенка неправильно, неполноценно, женщина наносит ему непоправимый вред. При острой недостаточности или избытке какого-либо вещества возможно рождение ребенка с видимыми или скрытыми (проявляющимися в различных условиях среды) аномалиями развития. Заканчивая рассматривать виды отягощенностей новорожденных, вызванных хромосомными, генными (матричными) и «средовыми» факторами, необходимо отметить, что здесь упомянуты типы и случаи, наиболее характерные для многих индустриально развитых стран с достаточно хорошо поставленной статистической службой.Алкоголь и врожденные аномалии
С давних пор известно, что алкогольные напитки оказывают пагубное влияние на потомство. В последние два десятилетия интерес к генетическим аспектам алкоголизма значительно возрос, и объясняется это тем, что потребление алкоголя в мире все больше распространяется среди молодежи и особенно женщин. Что же необходимо знать каждому о действии алкоголя на его организм и о возможных последствиях этого действия на грядущие поколения? Сейчас твердо установлено, что в общей популяции человека встречаются значительные индивидуальные различия в реакции алкоголя на организм. Одни индивидуумы не способны перерабатывать алкоголь, а другие, напротив, могут и даже в больших дозах. Основу такого различия обеспечивает локус (блок генов), образующий фермент алкогольдегидрогеназу. Иными словами, различная реакция людей на потребление алкоголя определяется генетически. У некоторых людей этот локус отсутствует, и они не могут принимать даже минимального количества алкоголя, исчисляемого единицами граммов. Доказано, что алкоголь и продукты его распада (ацетальдегид) нарушают синтез белка на уровне образования РНК (транскрипции) и на уровне сборки белка на матрице РНК (трансляции). Известно также, что алкоголь и его метаболиты даже в небольших дозах могут нарушать целостность хромосом, то есть вызывать хромосомные перестройки, а значит, и уродства в потомстве. Алкогольные напитки в том их разнообразии, в котором они имеются в настоящее время, следует рассматривать как сложные смеси, компонентами которых, помимо этанола, являются различные спирты, альдегиды и эфиры, куда входят сивушные масла и другие органические и неорганические вещества, способные также отрицательно влиять на обмен веществ в организме. Употребление алкоголя беременными женщинами имеет особенно тяжелые последствия — мертворождения, недоношенность, гибель детей в первые недели жизни, различные врожденные уродства или тяжелые, зачастую необратимые, внутриутробные поражения центральной нервной системы (разные формы алкогольной эмбриопатии). Алкоголь — важнейший тератогенный (повреждающий) фактор, действующий на плод и прямым, и косвенным путями. Тяжелые формы эмбрионального «алкогольного синдрома» встречаются в США с частотой от 1 на 880 до 1 на 2400 новорожденных. Клиническая картина этого синдрома определяется гипотрофией, черепно-лицевыми деформациями, соматическими уродствами, повреждениями мозга, включая нарушения психомоторного и интеллектуального развития. Следует отметить, что не у всех детей с эмбриональным «алкогольным синдромом» развивается полная картина болезни, то есть не все перечисленные отклонения могут встречаться у одного ребенка. Многочисленные литературные данные по ряду стран свидетельствуют чаще всего о нарушении общего развития, уменьшении массы новорожденных, изменениях в нервной системе и ухудшении умственного развития детей. Но главная тяжесть эмбрионального «алкогольного синдрома» ложится на психику и интеллектуальное развитие. Судьба детей с тяжелым «алкогольным синдромом» зависит главным образом от степени поражения нервной системы. Ученые отмечают, что более чем у 50 % больных наблюдается задержка психомоторного развития независимо от условий, в которых они живут в детстве. Важно также заметить, что интенсивность «алкогольного синдрома» зависит от дозы алкоголя, употребляемого матерью в течение первых месяцев беременности, от концентрации в материнской крови этилового спирта и от стадии алкоголизма матери. В последнее время мир встревожен пагубным влиянием алкогольных напитков на наследственные качества человека, на здоровье последующих поколений. Во многих странах борьба с алкоголизмом возведена в ранг государственной политики, и наука всячески помогает этому. На Международном симпозиуме, посвященном «алкогольному синдрому» (1975 год), убедительные факты привела доктор Хансон. Она рассказала о результатах обследования в штате Вашингтон 1529 матерей и их детей. Оказалось, что у матерей, не употреблявших спиртные напитки или употреблявших их в небольшом количестве, рождалось лишь 2 % детей с отклонениями от нормы (лишние пальцы на руках и ногах, аномальные складки кожи на ладонях и аномальный разрез глаз, низко посаженные уши, врожденные пороки сердца и т. д.), у умеренно пьющих эта цифра увеличилась до 9 %, у сильно пьющих — до 74%. Необходимо подчеркнуть, что в последней категории, как правило, зарегистрировано не одно, а несколько отклонений от нормы у новорожденных. Наибольшее беспокойство врачей вызвал тот факт, что 12 % детей, родившихся от матерей-алкоголичек, имели размер головы значительно меньше нормы. Последнее часто свидетельствует об умственной отсталости ребенка. В группе совсем не пьющих и умеренно пьющих женщин подобное явление отмечено в десятки раз реже.О вредных привычках
В настоящее время во всем мире курение признано как одна из актуальных проблем, стоящих перед обществом. Во многих странах очень многие молодые женщины подвержены этой весьма вредной для организма привычке. Социологические наблюдения показали, что доля курящих среди молодежи до 25 лет гораздо выше, чем среди более старших категорий населения. Вред от курения настолько очевиден, что борьба с ним стала заботой государств. Тем не менее мало кто знает, что курение приносит громадный ущерб и для будущего поколения. Цитологические исследования показали, что курение (даже не интенсивное) может вызвать стерильность мужчин и женщин (отсутствие способности к оплодотворению), снижает массу тела новорожденных, приводит к гибели эмбриона и мертворождению. Курение влияет на некоторые биохимические показатели организма. Так, например, под действием никотина увеличивается концентрация магния в сыворотке крови, меняются картина периферической крови и активность ряда ферментов в клетках. Кроме того, увеличивается уровень фибриногена в плазме, что повышает вероятность заболевания ишемической болезнью сердца. Американские врачи пришли к выводу, что для женщин курение опаснее, чем для мужчин, так как оно является причиной почти половины всех случаев заболеваний коронарных артерий у женщин в молодом и среднем возрасте (у мужчин эта зависимость меньше). С помощью тонких биохимических анализов установлено, что под воздействием никотина сигаретного дыма происходит значительное торможение синтеза белка в мозговых клетках. Доказано, что никотин тормозит синтез белка и в клетках печени. За последнюю четверть века отмечено увеличение числа заболеваний раком легких среди курящих женщин. Как показали результаты исследований, жены курящих мужей заболевают раком в 2 раза чаще женщин, чьи мужья не курят. Значит, вредно не только само курение, но и вдыхание табачного дыма, то есть пассивное курение. Доказано, что вдыхание табачного дыма на 10—30 % увеличивает опасность заболевания некурящих раком легких. Пассивное курение вызывает у детей в возрасте до двух лет кашель и развитие пневмонии и бронхита. Думается, что эти убедительные неблаговидные факты дают основание для обсуждения вопроса о курении на семейном совете: продолжать курить или оставить эту вреднейшую привычку.Несколько слов о кофе. В последние годы появилось много работ, изучающих влияние кофе (кофеина) на здоровье человека. Начало этим работам было положено исследованием, показавшим увеличение заболеваемости раком поджелудочной железы у любителей кофе. Однако были опубликованы и материалы, «реабилитирующие» кофе. Канадские ученые называют одной из причин противоречивости получаемых разными авторами данных различное содержание кофеина В исследуемом кофе. Они считают, что реальное содержание кофеина необходимо определять более точно. Для установления роли кофе в возникновении раковых заболеваний необходимо прежде всего разработать надежные методические рекомендации для изучения влияния кофеина на здоровье, отсутствующие на настоящий момент. А пока бытует мнение, что потребление этого напитка в умеренных количествах здоровыми людьми (не предрасположенными к инфарктам, гипертонии, гормональным нарушениям) не представляет опасности для здоровья. Однако исследования в этой области продолжаются.
Феномен «старой первородки»
Для рождения полноценного потомства большое значение имеет возраст матери. По данным статистики, средний возраст невест в СССР в 60-х годах был значителен и составлял 26 лет (табл. 9). Почему же возраст невест так сильно увеличился? Причина здесь в том, что современному развитому обществу нужны образованные граждане. Обучение же в наш век научно-технической революции длится долго. Это — 10 лет школы, два-три года работы (часто) и, наконец, не менее пяти лет института. Реально ли обзаводиться семьей и детьми в студенческие годы? Здравый смысл подсказывает, что нет. Часто желание обзавестись семьей у молодых остается лишь мечтой. Необходимо подчеркнуть, что главным в период получения образования многие женщины считают равенство с мужчинами, экономическую независимость, материальный достаток. Практически в каждой стране, кроме Китая, женщин больше, чем мужчин, и поэтому некоторым девушкам вовремя обзавестись семьей просто не представляется возможным (табл. 10). А выйдя замуж, например, в 26 лет, женщина, естественно, только в 27 лет (с учетом благоприятных обстоятельств) может стать матерью. Женщин 25 лет и старше, рожающих впервые, медики называют «старая первородка». К этому времени женщина физиологически изменяется так, что вероятность различных осложнений во время родов значительно возрастает. (Особенно у «старой первородки» деформированы кости и хрящи таза). В таких случаях во время родов необходима повышенная готовность врачей, чтобы вовремя оказать помощь матери и ребенку. Таблица 9. Динамика возраста женщины, вступающей в брак [Урланис, 1963]| Возраст, лет | Удельный вес, % | |
|---|---|---|
| 1910 г. | 1960 г. | |
| До 20 | 54,5 | 26,3 |
| 21-25 | 31,0 | 40,7 |
| 26-30 | 7,3 | 11,6 |
| 31-40 | 4,7 | 10,9 |
| 41-50 | 1,9 | 4,2 |
| Старше 50 | 0,6 | 6,3 |
| Возраст, лет | Мужской пол | Женский пол |
|---|---|---|
| Новорожденные | 106 | 100 |
| 18-20 | 100 | 100 |
| 25-29 | 90 | 100 |
| 60-64 | 80 | 100 |
| 65-69 | 70 | 100 |
| 70-74 | 65 | 100 |
| 75-79 | 60 | 100 |
| 80-84 | 50 | 100 |
| Старше 80 | 45 | 100 |
Глава 5. Социальная среда и человек
Низкая рождаемость и как следствие — «эффект одиночки»
В настоящее время средняя рождаемость в мире невысока — 2 %. Эта цифра означает, что на тысячу жителей приходится 20 новорожденных. В СССР в начале 1980-х годов рождаемость была ниже среднемировой и составляла примерно 12 детей на тысячу жителей. В предвоенные же годы (1940 год) этот показатель был гораздо выше — 30 новорожденных на тысячу жителей. Из приведенных цифр видно, что детей в мире стало меньше, а, значит, родительской любви на каждого ребенка (особенно если учесть увеличение продолжительности жизни современного человека) поприбавилось. В результате успехов научно-технической революции увеличилось количество свободного времени и улучшилось материальное благосостояние родителей, что также положительным образом сказалось на воспитании подрастающего поколения. Однако для современных детей в СССР характерно и то, что до 16 лет они фактически лишены возможности участия в труде, так необходимого для правильного формирования будущих граждан. Но вернемся к родительской любви. Относительно ее выдающийся советский педагог А. С. Макаренко утверждал, что избыток этого чувства более вреден в воспитании характера будущих граждан, чем его недостаток. По его мнению, оптимальное число детей в семье должно быть три и больше при разнице в возрасте 1,5—3 года. В таких семьях формируется благоприятный для воспитания микроколлектив, идет «шлифовка» будущего характера, закладывается и вырабатывается его гражданская позиция. Такие качества, как эгоизм, эгоцентризм, получившие в мировой социологической литературе название «эффект одиночки», для этих детей не характерны. И много внимания и энергии придется затратить педагогам на воспитание детей, выросших в семьях без сестренок и братишек, пока все острые грани их поведения перестанут вызывать негативное отношение, а порой и осуждение окружающих. Итак, из вышеприведенного следует, что снижение рождаемости в свою очередь порождает такое негативное общественное явление, как «эффект одиночки», значительно усложняющее жизнь конкретного индивида и, конечно, окружающих его людей. Последнее, разумеется, не может не отразиться на общих нормах отношений между людьми, которые в целом характеризуют нравственность человека и общества.40 % интеллекта — до трех лет
Современными психологией, педагогикой, социологией и другими науками, изучающими вопросы формирования человеческой личности, установлено, что около 40 % интеллекта будущего гражданина формируется до трех лет, а остальные 60 % — в течение всей его последующей жизни. Первые три года жизни ребенка имеют чрезвычайно важное значение, так как именно в это время формируются основные критерии отношения будущего гражданина к окружающим его людям, а также к предметам, ценностям и в целом к миру. Известно, что двух- и трехлетний ребенок задает взрослым, окружающим его, в среднем около пяти — семи миллионов вопросов. Следует учитывать, что разные дети реализуют свою любознательность по-разному. Анализ показал, что случайных, ненужных вопросов у детей нет. Если на все поставленные малышом вопросительные фразы отвечать спокойно, доброжелательно, ровно, терпеливо, не отмахиваясь от его любознательной назойливости, то можно надеяться, что на его развитие оказано большое, но, естественно, не исчерпывающее положительное влияние. От трех первых лет зависит не только интеллект ребенка, но и в значительной степени будущее самих родителей — будут ли они счастливы с детьми или последние привнесут в родительскую жизнь огорчения. Поэтому понятны тревога и беспокойство и родителей, и ученых по поводу часто встречающейся в жизни вынужденной разобщенности детей и родителей в эти первые три года — столь важные для ребенка, родителей и общества в целом. И радость родителей, получающих место в детских яслях, конечно же, относительна. В этом учреждении в среднем на одного воспитателя приходится 25—30 детей, и задача его не в том, чтобы ответить на те самые пять — семь миллионов вопросов, умноженных на количество детей в группе — ибо задача эта нереальная. В детских яслях решаются вопросы куда более прозаические — вовремя накормить, одеть, уложить спать, сменить штанишки и т. д. О том, что в первые годы жизни ребенка можно потерять нечто большее, наверстать, вернуть, восполнить которое позже уже не удастся, мало кто задумывается. А ведь за эти годы ребенок в результате ограниченного общения со взрослыми недополучит так много того, что формирует его интеллектуальное развитие, потенциальные возможности, и в итоге реальный интеллект гражданина получится значительно обедненным. Отрадно, что это начинают понимать все большее число специалистов и родителей. Указанная проблема не могла пройти незамеченной и для СССР. В настоящее время в стране действует Постановление о предоставлении женщинам послеродового трехгодичного отпуска по уходу за ребенком (1,5 года оплачиваются). Это позволит более плодотворно решать вопросы, касающиеся воспроизводства полноценных, здоровых граждан с высоким интеллектуальным уровнем. Эта мера — шаг к уменьшению детского правонарушения в нашем обществе. А в идеале — искоренение такого правонарушения. Несомненно, возможность домашнего воспитания детей до трех лет повлечет за собой снижение возраста невесты, который в настоящее время в СССР составляет 24 года, что, в свою очередь, приведет к уменьшению количества новорожденных, отягощенных наследственными заболеваниями, и к общему увеличению рождаемости. Это чрезвычайно важно для страны, ее будущего. Матери получат возможность заниматься воспитанием своих детей в первые годы их жизни, что, непременно, плодотворно скажется на интеллектуальном и физическом развитии потомства.Глава 6. Любовь, инстинкт, рассудок, семья
Печальный факт свидетельствует, что дети есть далеко не во всех семьях. Установлено, что около 10 % браков в мире бесплодны. Еще около 20 % семей не могут иметь детей из-за спонтанных абортов и выкидышей. «Виновниками» бездетности в одинаковой мере могут быть и мужчины, и женщины, однако причины их бесплодия (как клинические, так и генетические) различны. В настоящее время описано несколько десятков наследственных болезней, или синдромов, сопровождающихся мужским или женским бесплодием. Спонтанные аборты у женщин могут быть вызваны целым рядом причин: нарушением хромосом плода или матери, несовместимостью плода и матери по системе групп крови АВ0, гетеро- или гомозиготностью по ряду заболеваний, влияющих на функции воспроизводства, многими наследственными болезнями матери, затрудняющими вынашивание плода в течение беременности и, наконец, различными клиническими нарушениями роженицы. Изучение спонтанных абортов показало, что около 20 % их связано с изменением кариотипа плода, а 5 % — с несовместимостью по системе групп крови АВ0 [Кулиев, 1974].Выбираем ли мы гены для своих детей?
Гены, которые имеются у родителей, могут перейти к детям только в половинном количестве (из 46 хромосом в половые гаметы попадают только 23). С этих позиций, если оба родителя гетерозиготны по фенилкетонурии, то в такой супружеской паре с вероятностью 25 % может родиться больной ребенок. Рассмотрим другую ситуацию. У одного из родителей 45 хромосом, но одна из двух 21-х хромосом присоединена к хромосоме 15-й. В этом случае с большой вероятностью может родиться ребенок с транслоцированным синдромом Дауна. Ни одна семейная пара не хотела бы иметь ребенка с наследственным заболеванием. Может ли генетика помочь таким родителям? Да, может. В разработке методов профилактики генных и хромосомных заболеваний (аномалий) современная генетика достигла больших успехов. В ее арсенале имеется метод амниоцентеза (рис. 24), с помощью которого благодаря биохимическим анализам и изучению хромосомного набора у клеток, которые слущиваются с плода и находятся в околоплодной жидкости, можно идентифицировать около 100 генных и практически все хромосомные отклонения, отягощенности у развивающегося эмбриона в период до 18 недель беременности. Метод амниоцентеза успешно используется в Японии. В этой стране в обязательном порядке и бесплатно для забеременевших пациенток старше 35 лет (читателю уже ясно, почему старше 35), а также для женщин, уже имеющих детей с отклонениями либо происходящих из семей с наследственно неполноценными родственниками, делается анализ околоплодной жидкости и находящихся в ней клеток от плода. При наличии наследственного заболевания у эмбриона пациенткам представляется возможность самим решать, рожать ребенка или нет. Такой подход позволяет в значительной степени снизить рождаемость наследственна неполноценных детей и в итоге ограждает семью от трагедии, а общество от необходимости организации специальных домов для детей-инвалидов. Метод амниоцентеза — относительно трудоемкая и дорогостоящая процедура. Однако экономисты США подсчитали, что стоимость анализа для 900 женщин намного ниже стоимости прижизненной госпитализации одного больного, которая оценивается более чем в 100 тыс. долларов. Ежегодно в США рождается около 4 тыс. детей с наследственными болезнями и их госпитализация обходится в 1,5 млрд. долларов в год. Рис. 24. Схема проведения амниоцентеза.
Рис. 24. Схема проведения амниоцентеза.
Из вышесказанного ясно, что родители должны тщательно следить за своим генеалогическим древом. Однако как узнать, гетерозиготен ли человек по данному летальному гену? Является ли он носителем транслокации? Наконец, кому делать амниоцентез? Ведь всем роженицам сделать такой анализ невозможно, да в этом и нет необходимости. Рождение ребенка с наследственным заболеванием возможно в тех супружеских парах, в родословных которых (одного или обоих супругов) уже встречались наследственные заболевания. Таким семьям следует обращаться в медико-генетическую консультацию, где врач по генеалогии родителей подтвердит вероятность рождения больного ребенка и при необходимости направит на цитогенетический (определение числа и формы хромосом) или биохимический анализ. Такие консультации в СССР открыты в Москве, Ленинграде, Киеве, Донецке, Минске, Алма-Ате, Ташкенте, Фрунзе, Ашхабаде, Риге, Вильнюсе, Тарту, Ереване, Кишиневе, Кемерове, Новосибирске и других городах. Анализ работы медико-генетической консультации при Институте медицинской генетики АМН СССР в Москве, проведенный Р. С. Патютко [1975], показал, что за 1973—1974 годы у 77 % семей, обратившихся в медико-генетическую консультацию, родились больные дети. Естественно, что родители не хотели и не ожидали появления больного ребенка и, обратись они в консультацию до зачатия плода, такого несчастья могло бы не произойти. Ученые США подсчитали, что более 5 % населения страны имеют наследственные отклонения и нуждаются в генетической консультации. Выявление гетерозиготного носительства и гена фенилкетонурии медико-генетическая служба проводит специальным тестом с пищевой нагрузкой фенилаланином (одной из аминокислот). А что же делать, если больной ребенок все-таки родился? Здесь на помощь приходит генетика. Установлено, что некоторые наследственные заболевания можно «лечить» на уровне фенотипа. В настоящее время возникло целое направление в генетике — «лечение» фенилкетонурии. Но как провести раннюю диагностику наследственного заболевания у новорожденного, если внешне у него не видно никаких отклонений от нормы? В этом случае медико-генетическая служба предлагает ряд скринирующих (скрининг — просеивание) методик. В настоящее время скринингу поддаются около 20 наследственных заболеваний у новорожденных. С этой целью у последних берется моча или кровь (из пятки) . Современное состояние генетики позволяет проводить скрининг одной пробы на разные наследственные заболевания в разных центрах, при этом для анализа высылается капля крови на бумаге в конверте.
Какая у вас группа крови?
Группа крови — врожденное свойство человека и неизменна в течение всей его жизни (онтогенеза). К настоящему времени известно несколько систем группы крови (табл. 11). Каждая из этих систем наследственно обусловлена. Невозможно найти двух людей (кроме однояйцевых близнецов), которые имели бы одинаковые группы крови по всем системам. Это явление используется в судебной медицине. В клинической медицине для переливания крови необходимо знание группы крови системы АВ0 (I—IV группы крови) и резус-фактора. Система групп крови АВ0 открыта в начале XX века австралийским ученым К. Ландштейнером при изучении поведения эритроцитов (красных кровяных телец) в сыворотке (жидкой части) крови разных людей. Ученый обратил внимание на тот факт, что эритроциты в сыворотке крови одних людей распределяются равномерно, а других — склеиваются. Используя разные комбинации эритроцитов и сывороток, он обнаружил три группы крови (I—III), а существование IV группы (более редкой) было установлено позднее. Частота встречаемости групп крови системы АВ0 в разных популяциях человека различна (табл. 12). Обладание одной из четырех групп крови определяется парой генов, пришедших по одному от каждого из родителей. Каждый ген может быть в одной из трех аллелей (функциональных состояний) — А, В, 0. Аллели А и В доминируют над 0, но, оказавшись вместе в одном организме, А и В проявляют совместное действие (кодоминирование) и обусловливают наличие IV группы крови (табл. 13). Многие считают, что у родителей и детей группа крови всегда одна и та же. Это заблуждение. Установлено, что совпадение здесь имеет место далеко не во всех случаях. Фенотипически (то есть биохимически, морфологически или другими методами) можно определить четыре группы крови: I(0), II(А), III(В) и IV(АВ). Фенотипы I и IV групп совпадают с их генотипами. Генотипы же ВВ и В0 (для III группы крови), АА и А0 (для II группы) без знания групп крови родителей различать невозможно. Таблица 11. Основные системы эритроцитарных антигенов *| Система | Год открытия | Основные аллели | Число аллелей в системе |
|---|---|---|---|
| АВ0 | 1900 | А, А1, В, Н | 8 |
| MNSs | 1927 | М, N, S, s, U | 18 |
| Р | 1927 | P1, P2, p, pK | 4 |
| Rhesus | 1940 | D, С, Е, с, е | 35 |
| Lutheran | 1945 | Lua, LuB | 17 |
| Keff | 1946 | К, k | 18 |
| Lewis | 1946 | Lea, LeB | 2 |
| Duffy | 1950 | Fya, FyB | 6 |
| Kidd | 1951 | Jka, Jkb | 3 |
| Diego | 1955 | Dia, Dib | 2 |
| Ii | 1956 | T, i | 3 |
| Xg | 1962 | X, ga | 1 |
Таблица 12. Частота встречаемости эритроцитарных антигенов системы АВ0 в разных популяциях человека, %
| Популяция | Число изученных людей | I(0) | II(A) | III(В) | IV(AB) |
|---|---|---|---|---|---|
| Коренное население Австралии | 603 | 54,3 | 40,9 | 3,8 | 1,0 |
| Голландцы | 14 483 | 46,3 | 42,1 | 8,5 | 3,1 |
| Население Южной Англии | 3 449 | 43,5 | 44,7 | 8,6 | 3,2 |
| Голландские евреи | 705 | 42,5 | 39,4 | 13,4 | 4,5 |
| Русские евреи | 1 475 | 36,6 | 41,7 | 15,5 | 6,1 |
| Бушмены | 336 | 83,0 | — | 17,0 | — |
| Венгры | 1 041 | 29,9 | 45,2 | 17,0 | 7,9 |
| Арабы | 2 917 | 44,0 | 33,0 | 17,7 | 4,1 |
| Японцы | 24 572 | 31,1 | 36,7 | 22,7 | 9,5 |
| Русские | 57 122 | 32,9 | 35,6 | 23,2 | 8,1 |
| Африканцы (Конго) | 500 | 45,6 | 22,2 | 24,2 | 8,9 |
| Китайцы (Кантон) | 500 | 45,5 | 22,6 | 25,0 | 6,1 |
| Венгерские цыгане | 925 | 28,5 | 26,6 | 35,3 | 9,6 |
| Индийцы | 2 357 | 30,2 | 24,5 | 37,2 | 8,1 |
Таблица 13. Наследование групп крови у человека
| Мать | Отец | ||||||
|---|---|---|---|---|---|---|---|
| Группа крови | |||||||
| I(0) | II(A) * | III(В) * | IV(AB) | ||||
| Группа крови | Генотип | Генотип | |||||
| 10 10 | 1А 1А | 1А 10 | 1B 1B | 1B 10 | 1A 1В | ||
| I(0) | 10 10 | 10 10 | 1А 10 | 1А 10 | 1B 10 | 1B 10 | 1A 10 |
| 10 10 | 10 10 | 1В 10 | |||||
| II(А)* | 1А 1A | 1А 10 | 1А 1А | 1А 1А | 1А 1B | 1А 1B | 1А 1А |
| 1А 10 | 1А 10 | 1A 1B | |||||
| 1А 10 | 1А 10 | 1A 1A | 1А 1А | 1А 1B | 1A 1A | ||
| 10 10 | 1A 10 | 1А 10 | 1А 1B | 1А 10 | 1A 1B | ||
| 10 10 | 1А 10 | 10 10 | 1A 10 | ||||
| 1А 10 | 1A 10 | ||||||
| III(B)* | 1В 1B | 1B 10 | 1A 1В | 1A 1B | 1B 1B | 1B 1B | 1A 1B |
| 1B 10 | 1B 10 | 1B 1B | |||||
| 1B 10 | 1B 10 | 1A 1B | 1A 1B | 1B 1B | 1B 1B | ||
| 10 10 | 1A 10 | 1B 10 | 1B 1B | 1B 10 | 1A 1B | ||
| 10 10 | 1B 10 | 10 10 | 1А 10 | ||||
| 1А 10 | 1B 10 | ||||||
| IV(AB) | 1А 1В | 1А 10 | 1А 1А | 1A 1A | 1B 1B | 1А 10 | |
| 1B 10 | 1А 1В | 1A 10 | 1А 1B | 1B 1B | 1А 1A | ||
| 1В 10 | 1B 10 | 1В 1В | |||||
| 1А 1В | 1A 1B | 1A 1B | |||||
Таблица 14. Связь групп крови с антителами и антигенами
| Группа крови | Антигены, содержащиеся в эритроцитах | Антитела, содержащиеся в сыворотке крови |
|---|---|---|
| I(0) | _ | Анти А, анти В |
| II(A) | А | Анти В |
| III(В) | В | Анти А |
| IV(AB) | АВ | — |
Таблица 15. Варианты несовместимости матери и ребенка (по группам крови системы АВ0)
| Мать | Ребенок | ||
|---|---|---|---|
| Группа крови | Генотип | Группа крови | Генотип |
| I | 10 10 | II | 1А 10 |
| III | 1В 10 | ||
| II | 1А 10 | III | 1В 10 |
| IV | 1А 1В | ||
| III | 1B 10 | II | 1А 10 |
| IV | 1А 1В | ||
Резус-фактор (Rh)
Система групп крови резус открыта в 1940 году К. Ландштейнером. Изучая кровь человека и животных, ученый обнаружил, что кровь примерно 85 % обследованных людей подобна крови обезьян резус. Было доказано, Что кровь этих людей содержит антиген, идентичный имеющемуся у резуса. Обнаруженный антиген был назван резус-фактором (Rh). Вскоре было показано, что присутствие (или отсутствие) этого гена наследуется. Рис. 25. Несовместимость по резус-фактору. Отец Rh+, мать Rh-, ребенок Rh+.
Рис. 25. Несовместимость по резус-фактору. Отец Rh+, мать Rh-, ребенок Rh+.
Резус-фактор может быть положительным или отрицательным. В первом случае он обусловливается доминантным (более сильным), во втором — рецессивным (менее сильным) геном. Если оба родителя имеют одинаковый резус-фактор (положительный или отрицательный), то иммунологического конфликта между организмом матери и плодом не происходит. Конфликт возникает, если отец имеет положительный, а мать — отрицательный резус-фактор. В этом случае плод будет иметь отцовский положительный (доминантный) резус-фактор, который вступит в иммунологический конфликт с материнским — отрицательным. Если же, напротив, у матери положительный резус-фактор, а у отца — отрицательный, то плод будет иметь материнский положительный (доминантный) резус-фактор, и в этом случае иммунологический конфликт не проявится. При иммунологическом конфликте между организмом матери и плодом первый ведет себя так, как будто это не его ребенок, а инородное тело. Антигены плода вызывают появление в организме матери антител, способных при высоких концентрациях (титрах) нейтрализовать развитие плода и освободить организм матери от «чужеродного» тела. Однако при первой беременности количество антител в организме матери не повышается до такого уровня, чтобы существенно повредить (или убить) плод: исход, как правило, бывает благополучным. Но при второй беременности к антителам, которые остались в крови матери от первого ребенка, добавляется еще определенное их количество. В этом случае эритроциты ребенка будут уже частично повреждаться. Если между первой и второй беременностями не было прерванных беременностей, то и второй ребенок рождается вполне здоровым. Но если до беременности производились переливания крови без учета резус-фактора, которые могут быть приравнены (в зависимости от количества прилитой крови) к беременности, то второй ребенок будет неполноценным (рис. 25). В Новосибирске и в других городах Западной Сибири отмечено несколько случаев, когда врачи делали (и неоднократно) переливание крови при тяжелых заболеваниях девочкам без учета резус-фактора (который был детально изучен лишь около 20 лет назад), В результате, когда девочки стали взрослыми, все беременности у них заканчивались гибелью эмбриона или спонтанным абортом. Однако современная медицина может помочь таким семьям иметь детей. Созданный в Ленинграде Институт переливания крови производит частичную замену крови матери с целью снижения титра антител до неопасных уровней для будущего плода.
Антигены лейкоцитов
Изучение системы антигенов HLA началось в 1954 году, когда был открыт первый антиген этой системы. К настоящему времени их уже установлено более 100. Широкий интерес специалистов к названной системе связан с тем, что она ответственна за явление гистосовместимости тканей. Более того, в 70-х годах была установлена связь между предрасположенностью к некоторым заболеваниям у человека и системой антигенов HLA. Выдвинуто несколько гипотез, объясняющих эту связь. В настоящее время доказана корреляция HLA со следующими заболеваниями: вирусным гепатитом В, хроническим гломерулонефритом, сахарным инсулинзависимым диабетом, диффузным токсическим зобом, рассеянным склерозом, раком, ишемической болезнью сердца и др. (табл. 16). Иными словами, HLA-антигены являются генетическими факторами риска. Следовательно, типирование населения по системе антигенов HLA способствует выявлению лиц с повышенным риском к какому-либо заболеванию. При этом представляется возможность проведения современных мер профилактики, а при необходимости и своевременного лечения.Таблица 16. Распределение антигенов (в %) системы HLA у больных с различной локализацией рака (I) и с ишемической болезнью сердца (II)
| I | II | ||||
|---|---|---|---|---|---|
| HLA-антиген | Здоровые (845) | Больные (124) | HLA-антиген | Здоровые (619) | Больные (202) |
| А10 | 19,64 | 29,03 ** | By | 18,1 | 52,2 |
| Aw19 | 12,78 | 7,25 | Ах | 17,24 | 16,83 |
| А28 | 8,04 | 3,22 * | В5 | 21 | 17,33 |
| Ах | 17,4 | 29,0 | В7 | 27,78 | 40,59 ** |
| В12 | 19,76 | 12,06 | В12 | 20,51 | 14,85 |
| В13 | 6,86 | 8,87 | В14 | 7,43 | 14,85 ** |
| В14 | 7,33 | 5,64 | В15 | 11,41 | 20,79 |
| В15 | 11,71 | 14,51 | В18 | 10,0 | 6,93 |
| В16 | 5,91 | 8,06 | В22 | 5,16 | 2,48 |
| В18 | 10,53 | 0,8 ** | Ву | 17,72 | 4,95 ** |
| В21 | 3,55 | 0 | Cw1 | 12,23 | 9,44 |
| Bw22 | 4,61 | 10,48 ** | Cw2 | 8,33 | 13,33 |
| В35 | 12,3 | 4,83 ** | Cw3 | 12,23 | 19,44 |
| В40 | 17,63 | 1,61 ** | Cw4 | 18,75 | 35,56 ** |
Генетический регистр
В практической медицинской службе на обращающихся за помощью к врачу принято заводить амбулаторную карточку, с которой, пожалуй, все знакомы. А вот что такое генетический регистр? Медико-генетическая служба, которая создана (и создается) в ряде стран, приходит к необходимости создания специальной карты — генетического регистра, содержащего объективные, постоянно дополняющиеся данные о лицах, имеющих генетические аномалии. Генетический регистр в каждом конкретном случае позволит судить о степени генетического риска. Создание генетического регистра диктуется прежде всего происходящими демографическими изменениями — снижением рождаемости, увеличением числа лиц среднего и пожилого возраста, уменьшением числа лиц детородного возраста, увеличением количества новорожденных с генетическими отклонениями и т. д. Ученые считают, что использование генетического регистра облегчит применение различного рода профилактических мер и позволит проводить учет их эффективности. Генетический регистр может быть полезен для целей планирования материальных средств на лечение, пенсионное обеспечение, содержание больных. Рассмотренный медицинский документ является своеобразной генеалогической картой родословной и особенно необходим врачу-генетику при консультировании молодежи, вступающей в брак.Генотип, болезнь, лекарство
Практически каждое пятилетие в мире издается каталог аутосомно-доминантных, аутосомно-рецессивных и сцепленных с полом (в половых хромосомах) наследственных признаков человека. И каждый раз список наследственных болезней человека увеличивается. С чем это связано? Новые болезни появляются в силу продолжающегося изменения генетического материала (мутационного процесса генов). Повышение наших знаний о биологии человека и совершенствование методов диагностики современной медицины также способствуют «расширению» спектра наследственных недугов. Клиническое описание многих наследственных заболеваний было дано в давние времена. Однако нельзя забывать, что в окружающей нас среде происходит постоянное увеличение таких мутагенных факторов, как радиация и различные химические вещества. Мутагенное действие радиации открыто еще в 1927 году, а химических веществ гораздо позже — в 1944 году, причем последние представляют даже большую опасность в связи с их огромным разнообразием и количеством, а также в силу повседневного и, казалось бы, незаметного действия этих веществ на целые популяции через окружающую среду — воду, пищу, воздух. Химические вещества действуют через изменение генетического материала (мутации) и экспрессии (степени выражения) существующих генов. Научно-техническая революция, расширяя производственную сферу человеческой деятельности и изменяя условия окружающей среды, поставила перед генетикой задачу определить оптимальные условия среды для определенного генотипа. К настоящему времени установлено более трех тысяч типов профессиональных заболеваний. Многие из них имеют генетическое предрасположение, поэтому описание генотипа полезно для профилактики профессиональных заболеваний. Знание генотипа имеет большое значение и для лечения больных. Например, не всем людям можно принимать некоторые лекарства в обычных терапевтических дозах, так как даже однократный их прием может вызвать тяжелые осложнения. Широкое применение различных средств лечения позволило выявить большое число наследственных изменений в активности ферментов (наследственных энзимо- и ферментопатий), участвующих в метаболизме лекарств и значительно изменяющих реакцию организма на лекарство: в одних случаях лекарство в обычных терапевтических дозах вызывает, в других — не вызывает лечебного эффекта (то есть бесполезно в процессе лечения). При отсутствии наследственных энзимопатий врачи довольно часто не отмечают терапевтического эффекта ряда лекарств, особенно при длительном их введении. Одна из причин здесь та, что многие лекарственные средства являются индукторами микросомальных ферментов печени, которые осуществляют инактивацию большинства поступающих в организм чужеродных соединений, в том числе и лекарств. При повышенной активности микросомальных ферментов печени некоторые лекарственные препараты в терапевтических дозах не оказывают нужного эффекта из-за быстрого выведения их из организма. Индукционный эффект лекарственных средств применяют в лечебной практике. Так, стимуляцию активности микросомальных ферментов печени используют с антитоксической целью для ускорения выведения канцерогенных полициклических углеводородов, азотистых красителей и многих других чужеродных для организма соединений. При лечении синдрома Кушинга активизирующее воздействие на микросомальные ферменты печени оказывает выраженный терапевтический эффект. Фенобарбитал как индуктор микросомальных ферментов печени используется при лечении физиологической желтухи новорожденных и болезни Жильбера. Пониженная активность микросомальных ферментов печени обусловливает замедленное выделение некоторых лекарств из организма и пролонгирует их действие. Активность микросомальных ферментов печени детерминируется генетически. Установлено, что по скорости метаболической элиминации людей можно разделить на три труппы: быстро, средне и медленно окисляющие лекарственные препараты. Естественно, что при назначении больному лекарственного средства, метаболирующегося микросомальными ферментами печени, врачи хотели бы учитывать активность этих ферментов. В настоящее время показано, что активность микросомальных ферментов печени у детей выше, чем у взрослых, то есть лекарственные средства у последних выводятся в 2,5 раза быстрее, чем у первых. У пожилых людей (60—85 лет) отмечается снижение активности названных ферментов. Некоторые лекарственные препараты противопоказано применять в определенных возрастных группах. Так, антибиотик тетрациклин Комитет по лекарствам Академии педиатров США не рекомендует назначать детям до 8 лет. Использование этого лекарства детьми до указанного возраста вызывает желтое окрашивание эмали зубов, ингибирование роста костей, гастероэнтериты и другие негативные для организма явления. Комитет по лекарствам Академии педиатров США подробно рассмотрел заболевания, при которых назначается тетрациклин, и пришел к выводу, что в 80 % случаев это средство можно заменить не только менее токсичным, но и более эффективным лекарственным препаратом. В связи с тем что в СССР в 1975 году издан перевод книги Л. Полинга «Витамин С и здоровье», а также напечатано несколько статей на эту тему, употребление витамина С (аскорбиновой кислоты) в стране получило широкое распространение для предупреждения простудных заболеваний. Однако хорошо известно, что чрезмерные дозы витамина С обусловливают образование оксалатных камней. Благодаря достижениям медицины в СССР ликвидированы многие болезни. И все-таки в среднем каждый человек один раз в два года болеет гриппом, а дети поражаются простудными заболеваниями гораздо чаще. Каждый ребенок переносит хотя бы одно инфекционное заболевание: корь, ветряную оспу, паротит (свинку), скарлатину. В то же время некоторые люди вообще не болеют гриппом. В детских коллективах можно встретить крепышей, вызывающих зависть у большинства мам — эти дети при прямом контакте с носителями инфекции не заболевают. Что это? Устойчивость некоторых генотипов к определенным инфекционным заболеваниям? Генетики считают, что такая устойчивость, действительно, возможна. Иммунная система человека «начинает работать» при инфицировании его вирусами или микроорганизмами. Оказалось, что иммунная ответная реакция организма на инфицирование контролируется генетически и в определенной мере связана с HLA-фенотипом. Однако здесь необходимо учитывать и средовой (главным образом пищевой) фактор. Недостаточное или несбалансированное поступление в организм белков, витаминов и микроэлементов приводит к повышенной чувствительности его к инфекции. В специально поставленных опытах по заражению животных вирусом ньюкаслской болезни отмечалось увеличение смертности среди животных, получавших пищу как с недостаточным, так и с избыточным количеством белка по сравнению о таковой среди животных с нормальным содержанием белка в рационе. Таким образом, в настоящее время перед медицинской генетикой наряду с исследованием факторов, присущих специфическому возбудителю, со всей остротой встала задача изучения наследственной и средовой компонент (сторон) возникновения инфекционных заболеваний.Генная инженерия — человеку
Заканчивая рассказ об основах генетики человека, наследственных заболеваниях и профилактике их, следует сказать о новых направлениях в лечении генных заболеваний с использованием методов генной инженерии. Это направление медицинской генетики возникло совсем недавно, однако его перспективность не вызывает сомнений. Так, исследования последних лет продемонстрировали возможность эффективной работы генов человека в бактериальной клетке. Генноинженерные работы позволили создать бактерии, которые вырабатывают интерферон — блокатор вирусов, гормон роста, инсулин и многие другие препараты. Эти исследования позволили в ряде стран начать широкую практическую наработку новых лекарств. С помощью генной инженерии получен целый ряд биологически активных веществ для медицинских целей. Так, синтезированы белки, регулирующие иммунный ответ, вещества с антивоспалительным эффектом или стимулирующие рост и развитие кровеносных сосудов и др. В настоящее время выделены несколько сот белков и установлена их первичная структура — последовательность аминокислот, а через генетический код синтезированы (собраны) их генетические матрицы. И нет сомнений, что в ближайшие годы с помощью генной инженерии будут получены многие лекарства направленного действия. Конечно, нельзя забывать, что «исправление», или «ремонт», генов у человека имеет два аспекта — технический и этический. Пока приоритет в развитии принадлежит первому. Специалисты доказали, что технически работы по генной инженерии реально выполнимы. В качестве убедительных доказательств последнего приводятся примеры успешных экспериментов на животных — мышах, крысах, коровах, овцах и обезьянах. Показано, что у этих животных в части клеток могут работать «отремонтированные» гены. Технология получения последних такова: на стадии, когда зародыш состоит всего из нескольких клеток, его извлекают (чаще всего вымывают) из организма матери; затем клетки разъединяют и в ядра каждой изолированной клетки вводят гены-матрицы, ответственные за синтез нужного признака или свойства. В результате получаются клетки с направленно измененным геном. Такие клетки возвращают в организм матери, и зародыш продолжает развиваться по обычной схеме, однако на свет появляется животное с «запланированными» свойствами. Уже сейчас в ряде лабораторий ученые получили мышей размером с крысу. В этих экспериментально созданных особях «работает» ген, синтезирующий крысиный гормон роста. Выведены такие коровы и овцы, в молоке которых содержится большое количество интерферона — блокатора вирусов, ингибитора трипсина — вещества, необходимого для хирургии, активатора плазмогена и многие другие ценные лекарственные препараты. И хотя этическая сторона генной инженерии пока остается проблематичной, практически это направление генетики вплотную подошло к медицине.Глава 7. Общество: проблемы, будущее
Национальность, язык, культура
Как известно, человечество состоит из трех рас — черной, белой и желтой, между которыми существуют всевозможные переходы. Отдельная раса, в свою очередь, состоит из множества наций. Примерно 20 лет назад Комитет по национальностям при Организации Объединенных Наций провел интересный эксперимент — исследовал структуру талантливости основных наций планеты (выборки составляли более 1 млн чел.). Оказалось, что ни одна из них не имеет каких-либо преимуществ по этому признаку. А поэтому ни одна из наций не имеет никаких оснований претендовать на приоритетное положение среди прочих народов. По логике, превосходных рас также нет. Необходимо подчеркнуть, что все нации и народности не являются «чистыми» да и не могут таковыми быть, так как «чистота», благодаря накоплению в матрицах летальных и полулетальных генов, давно привела бы к вымиранию народов. Доказано, что коэффициент смешения наций довольно значителен и имеет устойчивую тенденцию к увеличению. По прогнозам генетиков, пройдет всего 40 поколений (считается, что у человека поколение в среднем длится 25 лет) и все нации смешаются. Человечество почернеет и прищурится. Это произойдет благодаря трем меланиновым генам, ответственным за темноту кожи, и двум генам, ответственным за узость глаз. Эти гены доминантны, они характерны для черной и желтой рас, которые в количественном отношении преобладают над белой и имеют тенденцию к дальнейшему численному росту. В итоге — европейский фенотип будет все более терять свои характерные черты. При смешении наций (а значит, и огромнейшего разнообразия фенотипов) вероятность гомозиготности (одинаковости генетических матриц отца и матери) значительно снизится, что, в свою очередь, снизит вероятность наследственных заболеваний. Здоровье человечества улучшится. Для иллюстрации сказанного приведем несколько примеров. Автору данных строк удалось услышать очень поучительный рассказ врача-якута о том, как 78-летний вождь племени небольшой народности (около 1000 человек), кочевавшей в начале 70-х годов нашего столетия по северо-восточному побережью Северного Ледовитого океана, пришел к заключению о необходимости смешения его народа с другими нациями («Нам нужен прилив чужой крови!»). За последние 10 лет в его племени, жившем в изоляции с другими народами, умерли все родившиеся дети. Старик не кончал университетов, но его мудрость позволила найти правильный путь к спасению племени. В книге Штерна «Основы генетики человека» приведен пример о евреях, выехавших более 100 лет назад из России и Польши в США и Канаду и в силу строгого соблюдения при вступлении в брак трех незыблемых для них принципов — принадлежность жениха и невесты к одной нации, к одной религии, кастовость — имеющих среди потомков высокий процент летальных заболеваний. Третий пример. В Японии по существующему законодательству отец, выдавая дочь замуж, должен выделить новой семье участок земли. Чтобы «не распылять» семейного землевладения, часто женихов и невест подбирают среди родственников. Но природу обмануть нельзя, и в таких семьях наблюдается резкое повышение числа наследственных заболеваний. Не напрасно издревле близкородственные браки были нежелательны. Люди из наблюдений знали — в таких браках полноценных потомков не будет. А в настоящее время практически во всех странах близко-родственные браки запрещены законом, не освящаются церковью.Итак, в мире происходит неизбежный процесс смешения наций. Конечно, признаки, присущие конкретным нациям, в этом процессе будут иметь тенденцию к стиранию. Однако широчайшие сочетания огромного количества генов обеспечат появление новых признаков. Иными словами, на смену одним признакам придут другие, одно качество перейдет в другое. (Вспомним; все течет, все изменяется.)
Язык — свойство нации. Язык — средство общения. При стирании граней между нациями логично говорить о стирании граней и между языками. Уже сейчас в каждом национальном языке присутствует очень много слов «чужого» происхождения. С успехами научно-технической революции в мире появляются новые термины, и словарный обмен между нациями усиливается. По-видимому, можно предполагать, что в будущем человечество будет иметь один язык, созданный на основе языка нации, задающей тон в общем вкладе в мировую цивилизацию. Кстати, математику, музыку (в том числе нотную грамоту), живопись можно считать своеобразными языками международного общения — они одинаково понятны всем нациям и обеспечивают обмен высокоинтеллектуальной информацией, чувствами — так называемой тонкой материей. Несколько слов о культуре. Культура, как и язык, динамична. Она развивается как вглубь — на основе традиций, обычаев наций, так и вширь — впитывая в себя лучшие духовные ценности разных наций. Процесс сближения культур обусловливается и расширением межнационального обмена во всех областях современной жизни. Наконец, при сближении наций культуры как свойства нации также испытывают взаимопроникновения. Итак, смешение наций — во имя избежания вымирания человечества — необходимо. С другой стороны, смешение наций и обусловлено тем планетарным процессом возрастания энтропии, то есть усреднения характеристик объектов среды, коим охвачены все виды материи на Земле, в том числе и человек. Плохо это или хорошо — это уже особый вопрос. И разговор здесь должен быть другим. А для природы такого вопроса не существует. Однако для нее характерна истина: природа не терпит равновесия, но стремится к равновесию.
Безопасность среды обитания
Мировой опыт показал, что созданные человеком и применяемые в больших масштабах в народном хозяйстве пестициды наносят всему живому на Земле огромный, пока в полной мере неоцененный, урон. Основной причиной пагубности этих ядохимикатов является общность строения их клеток и генетических программ всей живой материи — от одноклеточных организмов до человека. Ранее мы указывали, что генетическая матрица всех видов живой материи состоит из одних и тех же четырех оснований, входящих в ДНК, а отличие по конкретным видам заключается только в различной очередности этих оснований в определенных генах, контролирующих признаки. Более того, отдельные гены (около 1000 пар нуклеотидов) кишечной палочки по очередности оснований абсолютно идентичны с геном человека. На протяжении многих миллионов лет природа тщательно отбирала наиболее универсальные генные блоки, и эти «заготовки» одинаково эффективно используются как у бактерий, так и у человека. В таком случае, если гербицид, инсектицид, фунгицид, зооцид, дефолиант и т. д. вредны бактериям, грибам, насекомым и животным, то, исходя из позиций родства всего живого на Земле, этот пестицид не может быть безвреден и для человека. Все пестициды представляют собой физиологически активные вещества и в разной степени в концентрациях, применяемых в быту и производстве, являются блокаторами, ингибиторами, генетическими индукторами, мутагенами, канцерогенами и тератогенами. Попадая в организм человека и «встраиваясь» благодаря общности строения на клеточном уровне в его генетический материал, пестициды активно нарушают последний и таким образом наносят значительный урон здоровью живущих и последующих поколений. К этому следует добавить, что пестициды очень стабильны. Время их разложения весьма продолжительно и особенно в районах с коротким вегетационным периодом и в зонах сухих степей. Известны случаи, когда в хозяйствах вокруг г. Красноярска использовались гербициды на полях за 3—5 лет до выращивания здесь овощных культур (капусты, свеклы, моркови и др.). Анализ показал, что содержание пестицидов в овощах при уборке превышало ПДК в 5—15 раз. Другой пример: за три года до закладки плантации миндаля в Молдавии в поле под кукурузу по так называемой интенсивной технологии были внесены гербициды; в силу сухости почв ядохимикат в нужный срок полностью не разложился, что привело к гибели посадок ценнейшей ореховой культуры. С каждым годом количество вносимых в почву пестицидов в СССР возрастает. По данным известного эколога чл.-кор. АН СССР А. В. Яблокова, в 1986 году в СССР в среднем на гектар пашни вносилось по 2 кг пестицидов; по планам 1990 года это количество удвоится, А в районах хлопкосеяния масса пестицидов уже в настоящее время превысила 200 кг на гектар. Последнее является причиной того, что население этих районов во много раз чаще жителей других регионов имеет заболевания печени. Научный подход требует всестороннего изучения любого нового химического вещества перед тем, как внедрять его в народное хозяйство, в среду обитания человека. Для этого необходимо прежде всего изучить мутагенные (на генном, хромосомном и геномном уровнях), канцерогенные и тератогенные свойства, а также период полного распада данного вещества. Необходимо сделать точные научные заключения о том, способно ли новое вещество повреждать наследственный механизм, вызывает ли оно рост раковых образований и, наконец, отравляет ли клетки и при каких концентрациях. Проверка показала, что на настоящий момент все без исключения применяемые в народном хозяйстве гербициды обладают большой мутагенной активностью. Мировая практика показывает, что степень изученности внедряемых в хозяйство пестицидов крайне мала. Например, в США только 10 % используемых ядохимикатов было проверено на мутагенную, 38 % — на канцерогенную активность и 40 % — на тератогенные свойства. В СССР положение в этом плане еще хуже. Лаборатории Госкомгидромета СССР из 500 применяемых в народном хозяйстве пестицидов способны контролировать лишь менее 25. К тому же фактически ни в одной стране пестициды не проверяются комплексно — на мутагенный, канцерогенный и тератогенный эффекты, причиной чего является достаточная сложность и трудоемкость таких анализов. Дело осложняется тем, что комплексный анализ необходимо одновременно проверять на бактериях, растениях и культуре тканей человека.В последнее время появилось очень много информации об опасности для здоровья человека таких химических препаратов, как нитраты и нитриты. В чем суть этой проблемы? Во-первых, в сельскохозяйственное производство с каждым годом поступает все больший ассортимент минеральных удобрений. Установлено, что опасность внесенных нитратов и нитритов тем больше, чем меньше они сбалансированы с другими элементами питания — калием и фосфором. Отсюда следует, что удобрения вносить нужно комплексно, зная запас разных элементов питания. Последнее, как показывает практика, соблюдается, к сожалению, далеко не в каждом случае. Во-вторых, нитраты под воздействием микрофлоры кишечника превращаются в нитриты, а последние, в свою очередь, взаимодействуя с гемоглобином крови, переводят железо из двухвалентного в трехвалентное состояние, свойственное метгемоглобину, не обладающему способностью переносить кислород и, таким образом, сильно затрудняющему тканевое дыхание. В-третьих, избыток нитратов приводит к образованию нитрозоаминов — сильных мутагенов и канцерогенов, об огромном вреде которых для организма говорилось выше. Как видим, внесение азотных удобрений без предварительного анализа почв и точного знания запасов калия и фосфора может иметь последствия весьма плачевные. А можно ли снизить негативное воздействие на организм рассмотренных выше ядохимикатов? Безусловно! Это — применение научно обоснованных севооборотов; прекращение внесения в почву вместе с органическими удобрениями сорняков (склады навоза, как правило, «питомники» сорняков); использование биологических методов борьбы с вредителями сельского хозяйства; наконец, применение сортов со специальными генами устойчивости к вредителям и болезням. В конце 1988 года в Москве состоялось Всесоюзное совещание, посвященное перспективам развития генетики в СССР. На этом совещании автор данных строк предложил свои принципы оценки генетической и токсикологической опасности факторов окружающей среды и научного подхода к установлению ПДК и радиационной активности, способные реально описать экологическую ситуацию. С учетом новых принципов ПДК пестицидов, нитратов и нитритов должна определяться не «на глазок» (как это имеет место в настоящее время), а исходя из экспериментальных данных, полученных на бактериях, растениях и культуре клеток человека. При этом ПДК пестицидов должны быть на порядки ниже концентраций, вызывающих достоверное повышение мутагенного, канцерогенного и тератогенного эффектов. Именно такой принцип необходимо соблюдать при установлении ПДК, чтобы обезопасить всех людей, значительно различающихся по своей «норме» реакции на разные химические вещества. И конечно же, кроме заботы о собственном здоровье, человечество должно помнить о среде обитания грядущих поколений. Оставить эту среду потомкам экологически чистой — благородная задача современных жителей планеты.
Последние комментарии
5 минут 35 секунд назад
22 минут 39 секунд назад
43 минут 24 секунд назад
3 часов 25 минут назад
10 часов 48 минут назад
16 часов 32 минут назад